Al righty welcome everyone so today we're going to be talking about single cell genomics um on Tuesday we basically talked about uh gene expression analysis and then I told you oh and there's a lot of complications with single cell genomics and we're going to talk about them in lecture 13 well guess what we're going to talk about them today because your problem set has basically the entire pipeline of single cell analysis Differential analysis pathway enrichments some single cell epigenomics uh all Blended in in an awesome Jupiter notebook why are we doing this so that as
you start thinking about your final projects you have all of the tools in your disposal and a lot of people have talked about single cell genomics in their projects so we want to enable uh and Empower all of you guys to be able to actually go and carry out all of these single cell analysis which are Frankly a lot of the frontiers of where biology is right now so um a lot of mysteries of how the Gen works and how disease impacts the body is very difficult to uncover when you're looking at bulk analysis however
the moment you're able to separate the different cell types in the body Things become much more um amendable to these types of analysis so let's uh Dive Right In so we're going to basically now start with why single cells and uh second make sure That everything is here good go why single cells basically what are the traditional approaches and uh sort of the emergence of single cell RNA sequencing across different types of Technologies because understanding the technological underpinings can also help you understand some of the issues with noise and um when the technology doesn't go
exactly right then we're actually going to dive into the bi ology first What are the questions that you can ask about single cells and we're going to focus on a lot of the work that our lab is doing on understanding the brain in the context of Alzheimer's in context of schizophrenia and so so forth then we're going to step back and say okay what are the basic computational tasks associated with single cell analysis and then we're going to look at some of the frontiers of the field of some deep learning representation learning methods for Understanding
single cells and then we're going to touch briefly into Beyond transcriptomics into some of the epig genomics multiomics and so so forth was excited yes awesome great let's do it so why single cell uh again there's many ways oh yeah go ahead the SCE is locked oh we'll release it after lecture uh yeah they're slowly you know what here that's why I always bring a thumb drive because it starts sinking up there and then uh it continues sinking until You know the end of days or at least the end of lectures um but um you
know there's also PDF being generated so the PDF might actually appear at some point but we'll see okay um I love how there used to be a time when the teacher would basically just write on the board and you would sort of see what the next is when it's written and now there's like oh gosh the slides are not yet online what are we going to do uh so it's uh you should be expecting more and that's That's the progress of technology and that's you know we should always be asking for uh you know any
kind of information that can be share should be shared and that's why recording the lectures for example you know there students on Harvard who are like you know back to back unable to come they're able to watch them online and and the first lecture got what is it a thousand views or something like that or 4 th like Anyway I posted it on YouTube and um there's like thousands of people who are watching it instead of talking about it which is great so so um it's kind of cool but anyway you guys are the originals
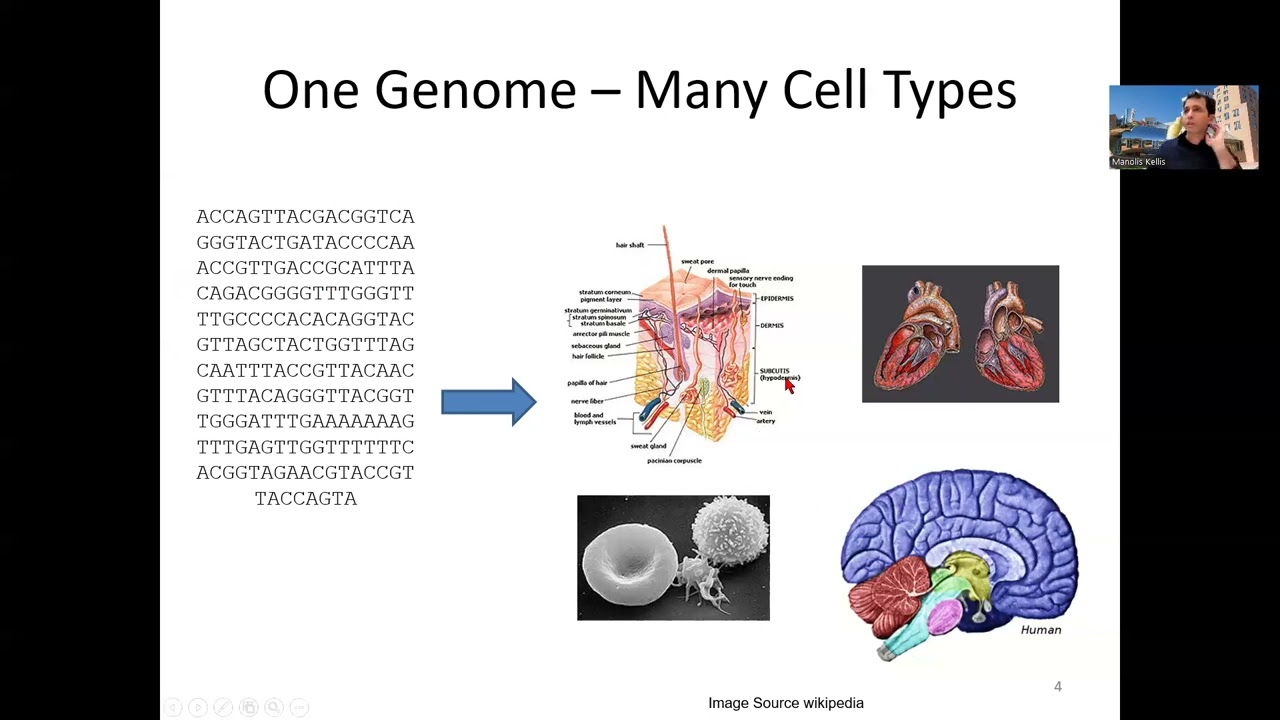
this is great this is a you know you get toour head start on on sort of frontiers of research um okay so uh yeah um there's many ways to think about um gene expression analysis the easiest and the analogy that has been used in many many uh talks and and lectures and Uh newspapers articles is the Smoothie versus the uh individual fruits versus the fruit salad or the the tart so so uh when you're doing bulk transcriptomics when you're basically taking a piece of liver and profiling it remember in the first lecture or yesterday's Tuesday's
lecture I was basically saying oh you look at brain expression versus liver expression it sounds reasonable right like if I tell my parents oh yeah we looked at Brain expression Rel liver expression makes complete sense to them but to a scientist especially somebody working on liver or brain they're like liver okay which of the 47 cell types in liver and somebody working on brain like which of the 143 subtypes of you know excitatory neurons in the brain Etc so so basically when you think about liver expression what you basically do is that you take this
extraordinary diversity of self types and you blend them together In a beautiful smoothie and then you're looking at the liver smoothie or the brain smoothie or the lung smoothie Etc when in fact it's a terrible analogy um when in fact the individual uh fruits are basically what you're after and and what we're going to look at today especially in the context of self- projected phenotypes a meure that and a method that we've been developing in my group with Geral Bond and others is that Even within cell types there are continuums of variation that are very
often biologically driven so even at the level of sorting the beautiful kiwis there's no such thing as like one type of kiwi there's still a a gradient of kiwi and the they're associated with different biological properties sometimes different disease properties and so on so forth so when our group started looking at single cell biology It was very very diff difficult to go back and we've kind of gone all out and and we've profiled more single cell than we could have imagined and that's what I'm I'm here to share with you today that basically if you
look at sort of the number of cells that people have been profiling it's now in the millions and from my group alone we have more than 20 million cells that we have profiled which sounds unfathomable basically if Somebody told you oh I'm going to you know give you 20 like remember when I showed you the gen expression by condition it was like thousands of genes hundreds of conditions now we're not talking about th hundreds of conditions or thousands we're talking about not t tens of thousands but like millions and tens of millions of conditions which
are the cells okay so that's why it's important because in the brain or in the lung or in the liver or in every cell Type in your in every tissue in your human body there's an extraordinary diversity of interplaying cell types and that's concept number one concept number two is that very often we're going to be talking about single cell but in the brain the neurons are sometimes extending all the way out to our toes okay so how do you do single cell profiling you're going to have to sort of pull out all of that
neuron take all of the RNA from all of the different Axons and dendrites and sort of you know unweave it and then profile it that's very hard and especially in the brain where the cell types including on nees asites you know and of course exidor inhibit neurons they're extraordinar interweaved with each other so it's very difficult to sort of pull up all of the parts and then sequence the entire content of the RNA of one cell so an approximation that we're always nearly always doing is single nucleus Sequencing because the RNA that's in the cytoplasm
at some point needs to be produced from The genome and that is done in the nucleus so you can think of it as an oximation of what being produced right now rather than the actual AC molecules that are sort of producing proteins in the cytoplasm of these cells moreover as we start thinking about more and more resolution I wouldn't even want to know what's the total content of RNA Inside the a single neuron what I would like to know is how much are sitting in this part of the axent and that part of the action
in these dendrites in those dendrites Etc in other words the transport of RNA along the cell body to the different parts is also extraordinarily important and I think that's also technological Frontier so there are some purists that will basically say oh single nucleus makes no sense why are you doing this and there's Other pures that will say RNA expression makes no sense everything happens at the protein level and other pures that will say protein makes no sense everything happens at the pro translational modifications of the protein level and others say oh post transation modification of
the protein makes no sense everybody everything happens at the localization of the post transation modifications and the orientation of them Etc ET ET So my answer to all of Those purists is we got to start somewhere and we start where the data is we start with the Technologies we start with what's accessible right now and based on what we learn we help Advance the field and the Technologies and we continue pushing the frontiers of single cell profiling to more and more different types of modalities and Technologies and so and so forth that will um not
be possible if we didn't push every technology to its extreme Based on what's available today in other words yes we could we could have waited 10 years back in 1995 to basically say Okay single sorry um single molecule sequencing is is around the corner and uh long read sequencing is around around the corner and sequencing is very expensive right now why sequence the entire Human Genome now let's wait you know a decade or two for the technology to improve but the catch is that the technology will not improve Unless you do it with the current
technology to show the usefulness to drive Innovation to drive entrepreneurship to drive technological advances to drive the market and the need for these advances that will then feed back everybody with me here so so that's why we keep pushing even though we know it's an approximation so again single nucleus RNA sequencing is extraordinarily awesome gives us a tons of resights but we should all be Cognizant that there will be more as more Technologies become available but until they are we're going to keep pushing the frontiers of what is available sounds like good F awesome so
again I talked about the smoothie I talked about the individual fruits and then there's the tart which is the third analogy of spatial transcriptomics which we'll uh allude to a little bit here but then we'll also talk more when we start talking about Imaging analysis and so so Forth and that's basically when you look at the tissue itself where every single one of those cells is positioned rather than only looking at sort of these cells pulled out of their context so basically the uh single cell technology you should realize is extremely limited because it tells
you about yes all of the exelator neuron layer two three and so so forth but it doesn't tell you exactly where these are positioned and maybe maybe the ones that Are position in this part or that part or associated with I don't know vasculature or not are actually different from each other and that you can get from spatial transcripts everybody with me here awesome so um great so why single cells I think I got a lot into that uh and you know let's uh talk a little bit more about this so individual cells again uh
what I've been saying is why single cell types why studying the Kiwi Separately from the banana is extremely important but what's also important is studying multiple kiwis separately and multiple bananas separately and the reason is that when you look at cells you know in the body or in a plate they actually look very different from each other and when you look at them in microscope they have different morphologies they have different expression levels of individual proteins and one cell to the other even of the Same cell type can be dramatically different moreover if you look
at the blood differentiation you have extraordinary diversity and almost a Continuum of cellular identity as you go from the hematopoetic stem cells which are the stem cell progenitors of all of the different blood lineages the hematopoetic lineages and then you have this extraordinary diversity of cell types that we know have different functions and then of course there's the Diversity within the cell types themselves of each of the different functions so all of this needs to be taken into account moreover why are they different they're different because of a stochasticity so basically every cell has an
expression program but which subset of genes gets activated at any one point actually differs So based on the genes that get activated first even though all of them are supposed to be turned on you might get some that get Turned on First and others second Etc and some of that is stochastics how will the particular regulator wash through quote unquote the different regions of the nucleus and open up different parts of the genome to express different genes there's also stochasticity in how a signal that is intracellular will permeate a group of cells and how these
cells will respond so The receptors of a particular cell might receive more or Less of a given environmental signal that then causes a cellular State transition and that can actually lead to a lot of diversity even between individual cells and then lastly developmentally every cell has a program that starts from a single genome and then through series of Divisions and maternal deposition of gradients and also where a particular cell is relative to you know the side of implantation or you name it um all of that will lead to Changes that happen at every cell division
that basically say all of you know this lineage after this one cell division will be ventral and all of these lineage will be dorsal or all of these lineage will be anterior and all of that lineage will be posterior and there's series of decisions that get made after every one of those divisions that basically lead to different identities which are remembered by the epome that we're going to talk about Next week they're remembered by the epome and then they determine the responses of each of those cells to subsequent signals is everybody with me here yes
okay can I get a 543 to one for how well you're following let see some fives this is great awesome some fours any questions who feels like they're learning stuff yes awesome great beautiful um okay so when you start looking at individual cells and some of The initial methods were basically taking a tissue dissociating individual cells and then padding these cells and putting each of these cells in a separate well and then amplifying the RNA of that well and then sequencing that one well okay this is the original single cell profiling and then you you
got like 600 cells and it was extraordinary and what you could see is how these cells can be dramatically different from each other so you can Distinguish different types of cells different responses different uh stages of activation of those cells after one hour after two hours after four hours after six hours of a particular stimulation you can kind of see which subset of cells is uh you know responding in which way and what subset of genes are activated in response so this is the expression Matrix that I showed you in last uh time lecture everybody
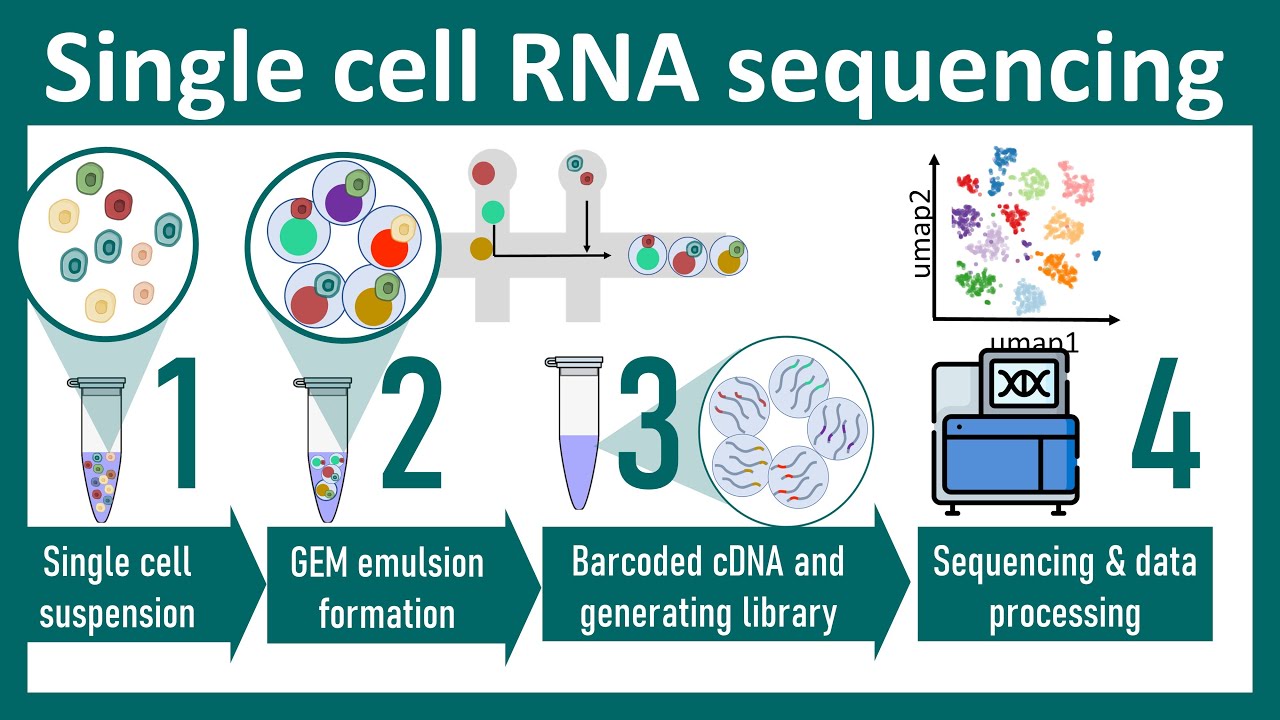
with me here so what we're Showing now is not hundreds of conditions but instead hundreds of cells all living in the same sort of organ or in the same sometimes animal or donor okay so that's the traditional approach of isolating individual cells using a pipeline then there's automated approaches for doing this where you're basically shooting cells through uh cell sorting apparatus that allows you to now start you know putting each of these individual cells in wellth and there are Technologies that allow you to now encapsulate every cell in a microfluidic droplet that allows you to
now capture individual cells in reaction Chambers that are not the size of a test tube that are not the size of a plate with 196 Wells or something but is instead the size of roughly one cell where basically those reaction Chambers are made out of little capsules little droplets that are encapsulating the cells in every one of those droplets you Can put a bead that contains millions of barcodes that will then tag every single one of these RNA molecules with the same barcode and then you can sequence the resulting RNA and look at the barcodes
and then try to infer which rnas came from the same capsule from the same droplet and therefore which rnas came most likely from the same cell everybody with me here and of course when we're isolating individual cells or when we're isolating individual Nuclei there might be stuff stuck on them and as we lice the nucleus or as we the cell there might be some ambient contamination of those cells and that contamination might be greater with the health of the original cell if you're taking cells from a cancer patient they might actually be more prone to
this type of contamination if you're taking cells from a very elderly individual or from someone with neurod degeneration as the neurons are dying the state of their Nuclei the state of their cells might actually be uh you know compromised not just just when you're carrying out your experiment but even before maybe there's more types of contamination that happen you know because that individual is sick or early and so so forth yes are these RNA molecules free mRNA or they slice so because we're looking at the nucleus it's both and you can you can basically get
At some of the Dynamics of gene expression patterns by asking what fraction of my mRNA molecules are actually unspliced versus spliced and there are techniques for building Cellular trajectories in a differentiation for example if you see that if you think that all of these cells are part of the same differentiation lineage leading from State a to State B and between State a and state B I need to express I Don't know 40 genes you could basically say oh let's find cells where this Gene is now fully spliced versus still unspliced Etc So based on the
coverage of the splicing of the you know introns in between the exons of every one of those cells you can actually tell a lot more by exploiting the nature of the data set but yes the answer is are going to be both spliced and unspliced because you're capturing nuclear RNA in many Other cases everybody with me here awesome good so again if you start looking at bulk and there's many cells that have little and many cells that have a lot you're going to mistakenly think that you know your tissue is somewhere in the middle when
in fact very very few cells are actually there in the middle similarly if you have some rare cells that Express a lot of given RNA and then you average a whole tissue you're going To think that oh every one of those cells has a little when in fact most have none and a few cells have a lot okay so these are some of the reasons and then this is now looking at at multiple genes on the x- axis so here you can see the actual Exon Inon structure so as you all know there's the original
genome here and then every tick mark is an exen a protein coding exen and then these exons get spliced as they you know get transcribed There's called transcriptional splicing and a maturation of the Mr that then gets exported fully spliced to the cytoplasm where translation begins so when you now sequence those RNA reads you will see that they fall on top of the protein coding Ain that's because in these experiments we're looking at cytoplasmic RNA okay in some cases you might see some reads in the intron and you might be wondering hm what is happening
here Could it be that this RNA molecule was not fully spliced could it be that there's um you know alternative splicing event and so so forth so that's the kind of thing that you should worry about basically if you look at now individual cells here every line is a different cell and then here every line is 10,000 cells okay and what you can see is that the individual cells when sequenc to sufficient depth with many of these early approaches you can basically see That they sometimes match exactly the 10,000 profile but other times what you
see is that the bulk sequencing of say 10,000 cells is dramatically different from some cells that just simply have no expression for that Gene everybody with me here so that I hope gives you an appreciation for these rare events that you know sometimes are happening basically this Gene is only expression a subset of cells and again when you look At Imaging you see the same thing reflected there this is not just a technical artifact but those genes that are uniformly expressed across cells when you stain them you see that they are indeed found in every
one of those cells but those that are only there in a subset of cells are indeed only there in subset of cells when you look at Imaging and those that are only in a handful are again only appearing in a handful so that Basically should reassure you that this is not just about Dropout events that this is really biological variability you could call it stochasticity or you could just simply call it variability statistics C that there's some kind of random process and yeah some of it might be random but what do you attribute to Randomness
is it that the receptor bound and then the cell randomly chose not to Respond or is it stasis because that that that um ligant didn't actually bind the receptor in some of those cells and then they responded in completely deterministic way to otherwise probabilistic environmental factors is everybody with me here but ultimately what what you need to understand is that the variability is there much of the time it is actually biological not technical but some of the time it will be technical some of the time it will be Stochastic because of the way that the
environment Works otherwise it will be stochastic just based on the way that the celles respond to deterministic event and of course most of the time it's going to be all of the above everybody with me here awesome who feels that they're learning yes awesome good um again you can kind of start asking about intercellular and you know variability between different cells and you can start getting a lot of insight As to what are some of those most variable or variable GS okay so now let's see how we went from capturing individual cells in some of
the earliest single cell papers or 10 cells or 100 cells to capturing millions of cells in the span of less than a decade which is mindball okay so what are the key technologies that gave rise to this dramatic uh Innovation so again if you look at the methods for isolating single cells you could just Simply pipet by eye individual cells and put them into 96 wall plates and then sequence each of the wells as if it was a sample from a different tissue or a different individual you could basically use capillary pipets under the microscope
to be able to sort of capture those better you can use this fact sorting that is throwing a laser that can detect the presence of cell surface markers or other protein markers in a collection of cells and as every cell Travels through that tube you can basically make a decision to keep it in the tube or outside based on this by applying a current or a magnetic field or you name it and that allows you to now capture a particular cell type that you might care about for example you might care about activ ated T
cells and there's a marker of activation that you can then sort for and gather them all together everybody with me here you could also use laser capture Micro dissection that is basically looking at a tissue and then capturing a tiny little part of that tissue cutting it with a laser and one of the most U widely used Technologies is the microfluidic technology that I mentioned earlier where you basically have cells from your suspension coming in one microfluidic tube then you have these individual beads that capture the barcodes that that have you know again thousands or
Millions of bar codes that all have the same code if you wish let's say you know a g g ttca for some cell and then t g a cc g g for another cell and then every time you see the exact same barcode you know that it was the same capsule and therefore most likely the same cell and then by adding these capsules with oil you're using the physics properties where these material and the oil are actually the lices buffer and the oil are not mixing therefore causing this Lices buffer to sort of Clump inside
one of these Chambers one of these bubbles that basically becomes your reaction chain everybody with me here and then there's uh you know other Technologies I'm going to talk talk about some of those okay so anyway the key the key idea is that the overall methodology is that you first isolate cells then you amplify the RNA and you can you know uh choose for example two populations of cells and you Can use microfluidics and then you effectively at the end do some kind of measuring or counting of RNA molecules and you can do that from
full length sequencing which basically will then give you the splicing patterns for each of those rnas or you can use that with what we call digital gene expression analysis that effectively only looks at the tail end of every RNA molecule why the tail end because when we sequence we use a Primer that initiates the polymer reaction through which we do the sequencing and that primer for a lot of eukariotic uh genes can be simply tttt because these genes finish with a a AA so there's a poly Aden that basically adds a bunch of A's at
the end of mRNA transcripts and then if you have the TT primer that allows you to now start sequencing the tail end of these genes and therefore capturing the number of times that mRNA Was expressed but not capturing the specific splicing pattern under which it was expressed everybody with me here who's following so far give me a 543 to1 beautiful that's awesome great all right so again there's many many different Technologies for single cell RNA sequencing there's uh you know different amounts of uh uh cells that are being captured there's uh different costs there's different
Sensitivity and there's different cell capture methods and you know you can kind of um look at this extraordinary diversity and you can see that for some methods you capture 50 to 100 cells or you know 500 to 2,000 5,000 to 10,000 then there's a cost per cell so basically when you're doing this uh traditional every cell is a well um approach coach you basically pay about $3 to $6 per sale which sounds ridiculously cheap unless you start Carrying about millions of sales and then becomes expensive there's pulled approaches that are basically looking at again you
know $3 to $6 per well and then there's these uh microfluidic based you know individual cell approaches where you basically have uh you know five cents to the cell which again sounds crazy okay so what are the key technology surrounding those again as I mentioned earlier you can basically use this laser Shooting and then these flowing of cells to basically choose individual cells based on these multispectral detectors and then put the cells of interest to the plate there's this microfluid chambers that I mentioned earlier where you're basically capturing and capsulating each cell in a well
and then that well that you know a micro reactor which is basically just a bubble you didn't have to pay with plastic for that Chamber has a bead that then has all of the same barcode on that bead and that barcode then gets attached to all of the RNA molecules of that cell so you basically capture the cell inside the bubble and then you lice the cell you release all of the mRNA molecules and all of these Mr molecules get the same tag okay the same barcode so this is what the bead looks like you
have a PCR handle you have a cell specific barcode and then you have Something that we call a unique molecular identifier a Umi what is that that is basically just a random chunk of nucleotides where you basically synthesize these nucleotides to just be random could be four it could be eight nucleotides which are basically just nnnnn which basically means any nucleotide why is that important because then you know that you're sequencing the exact same molecule that was mistakenly ified a thousand times so therefore if You if you see the same unique molecular identifier 100 times
it means that that's exactly the same RNA molecule that gets sequenced and therefore it's an artifact you shouldn't say oh there's a thousand copies of that Gene everybody with me here so the Umi is usually a measure of uniqueness and that's basically what we're going to you know when we say how many cells did I capture you'll say well how many or how many cells that's the number of bar codes how Many unique RNA molecules that's thei okay everybody with me here awesome so then the coolest part about this is that after I've done this
step of basically in my bubble tagging every RNA that came from that cell with the same barcode I can then blend all of the bubbles together and do one sequencing reaction so basically this entire process is just to to tag all of the rnas that came from the same cell or nucleus and then what I'm Actually sequencing is just a bulk sample it's the same thing as I did before okay only I did this magic to basically tag my rnas based on where they came from 543 to1 who's with me on this one yes beautiful
okay so that's that and then in the middle you have this very cool technology that I will explain in the next slide which is um that there's another way to do this which is you have your let's say million cells and in the first round you split Them into 100 Wells and all of the 100 in this sorry all of the cells in the blue well I just call her blue with a barcode that's like I don't know six nucle ties for example or however many nucleotides I need to distinguish let's say 100 okay whatever
L for of 100 is you then blend all of your cell together but now they've been colored blue or red or orange or green or yellow Etc everybody with me here because they have a unique barcode added to every RNA of That cell you then blend them all together and split them again you now add a second barcode on top of the first one and this could be you know pink or yellow or green or you name it some colors that you did not reuse before and because this is digital it's RNA so you're adding
individual barcodes then this can be completely different codes from that one everybody with me here and then you blend them again and then you add a third barcode so by the End of the exercise every RNA in every cell has gotten three different barcodes one from the first reaction one from the second reaction one from the third reaction round one round two round three if I have a 100 Wells here and 100 Wells there and 100 Wells there the chance that two of them the two mrnas will have exactly the same barcode but when through
different cells is effectively NE basically one in 100 Time 100 time 100 that's a million okay so I will basically have a million distinct possible paths through to three rounds of wells and three rounds of barcoding so then I can sequence my entire thing and effectively call all of the rnas that have exactly the same three barcodes as having gone through the same three Wells because they were attached to the same cell who thinks this is cool yes awesome Good and who's with me 54321 yes awesome beautiful okay so that's basically the you know the
third technology so the second technology is by far the most common and then you know this is the one where you're basically adding these barcodes and then you're dissociating the cells and the Sig whole reaction but this is I think you know just very very cool as well and again it sort of teaches you different ways of combining okay everybody with me on Technologies all right let's now talk about biology so basically again there's three parts to the lecture one is the technological part one is the bi biological part and one is the computational part
okay so we talked about the technological part let's now dive into the biological part what do we actually do when we have tons of single cell data okay and this is a lot of research that happens in my lab and it's kind of fun because you know I get to Teach about stuff that didn't exist a year or two or three ago because we you know we just built it so let's see what do we do so basically what's the goal of our lab the goal of our lab is to understand the circuitry underlying disease
to basically look at how are genetic differences or environmental differences leading to molecular differences that then manifest in disease States those molecular differences can Be at the RNA level or at the epigenome level and we're going to talk about epigenomics next week but then we profile massive numbers of both healthy and disease samples with single th profiling and then we carry out all of the stuff that I'm going to show you in the next section to find what are the driver genes uh regulatory regions Regulators cell types Pathways processes Etc and then we infer from
this analysis causality Because genetics gives us causality because we know that this genetic variant leads to differences in a causal fashion rather than those differences leads to the genetic variant okay because you inherit those from your parents and those you basically measure after a lot of environmental exposures it's very unlikely that this is in fact causing the genetic differences much more likely that the genetic differences are causing Whatever differences you see but of course the genetic differences could be then I don't know causing you to eat a lot of McDonald's and then you know the
the differences are not directly linked to that genetic difference they're linked to you know the fact that you had an unhealthy lifestyle okay um unless having salads at McDonald's which is also great we then try to infer these causal paths by understanding how are these Genetic variants acting at the molecular level to then figure out where to intervene okay so what I'm going to tell you now is some of the lessons we're learning for applying this to a lot of um disase samples so we have set up a large number of amazing collaborations applied for
for dozens of Grants from the NIH to fund each of those collaborations and we're basically studying dozens of different traits at single cell Resolution this takes an enormous amount of coordination across doctors who are providing the samples extraordinarily generous patients who are sharing you know their their samples postmortem and their families of course amazing Heroes of of these research all of the technicians in the lab who are basically Gathering these samples you know sorting profiling them then all of the experimental extraordinary technical expertise by the Experimental members of our team that are basically then dissociating
carrying out all the steps that we talked about earlier pushing very often new technologies forward to make all of these possible and then of course the computational analyst that then come in and analyze these data and of course the whole team together working to analyze interpret the results okay so that's you know it takes a village basically to to to create any one of these studies but What we basically do is that for Al disease or for frontal temporal dementia Louis body dementia ALS Huntington psychosis in the context of Alzheimer's psychosis in schizophrenia bipolar disorder
Down syndrome autism depression suicide PTSD Aging for all of these differences between controls and cases we basically then profile dozens of cell types across tens of millions of cells very often in multiple regions of the brain very often also in other tissues Outside the brain and then most of the time we do RNA profiling through single cell transcriptomics which we're talking about mostly today and in some cases also DNA accessibility which we're going to talk about much more next week everybody with me here so then what do we find so in one of the first
studies in the postmortem human brain and the first study of Alzheimer's at single cell resolution we basically gathered 80,000 cells from 48 individuals and this was not the beginning but the middle of a very long and extraordinary collaboration with uh Lee weai and her lab she's a director of The picar Institute for learning in memory uh H ready Mattis led a lot of these studies both experimentally and lately computationally and you know we've been U you know releasing a series of papers but that was the first paper by matis Jose DAV who basically now has
his own lab in Italy H matx has his own Lab at upit and what we basically looked at is what does this all mean what are you know what are all these sales doing okay so I'm going to show you a lot of the computational steps to figure this out but very briefly the first step is clean up the data and I'm going to tell you some some stuff about that what does clean up the data mean that basically means find metrics of quality that allow you to throw away individual cells or throw away a
whole Experiment or throw away individual genes that might be misbehaving and so so forth after you've done that you basically have this expression Matrix that I showed you in the last lecture you basically have a matrix of a lot of genes and a lot of cells everybody with me here the next after after step after that is to Cluster these cells together to basically figure out what are distinct cell types if you wish okay and this can be kind of challenging in some Cases why would you call this whole thing o progenitor cells and not
you know a subset of those okay in other cases you have excitatory neurons that are sort of Shifting across a whole gradient of layer 1 two 3 four five of the cortex which actually have distinct expression patterns but they're more of a Continuum than discrete cell types so calling individual cell types from data can sometimes be difficult okay so this is the first step basically clustering To identify what are the major drivers of variation and that's a form of unsupervised learning as we talked about in the last lecture you've now clustered your cells and at
some point you're going to call individual clusters and you're going to give them a name you're going to say well these celles here that have the same barcode cluster together and I'm going to guess that they they you know they have a particular identity how by looking at what are the genes That the most expressed in those cells and again we're not showing up here from you know the Stone Age we're showing up here with Decades of research of extraordinary scientists minutely studying the expression of individual genes in you know cells that they studied under
microscope you know individual cells were have always been around and and they've always been individually studied before these massive scale Methodologies okay so the difference here that we're looking at two million cells whereas the previous studies we're looking at 20 cells or 100 cells everybody with me here so with these 84,000 cells from 48 individuals we basically asked how does the expression pattern of those cells correlate with the phenotype of those individuals that's the first thing you want to ask so those 48 individuals are you know shown here as different columns And every person has
some phenotype associated with that person some people we're calling them controls or non-alzheimer's why because if you look at the phenotypic signatures of Alzheimer's at the pathology level they don't have them neur Tangles nft um pathology uh neuritic plaques amid Beta And so so forth you see these are very low value here but very high high value here is everybody with me here awesome then you can also look at signatures of Parkinsonism and signatures of cognitive decline and you can see here that the global cognition for those individuals seems to be high except for this
one person that shows some signs of peronism perhaps and then the uh individuals here have in general very strong cognitive decline and a lot of pathology and then there's another group here that is somewhere in the middle so we call this mild cognitive impairment or MCI or mild ad and we call those uh ad or severe ad And those non everybody with me here and then we basically ask you know what what is different between those people and we can basically now start studying individual genes that are differentially expressed between cases and controls so now
that you have all of these data sets you can now ask as a group are the cells that are coming from Alzheimer's individuals showing higher expression for example for these genes than the cells coming from Non-alzheimer's individuals everybody with me here now let's talk about statistics what statistical test would you use you basically have I don't know uh 20,000 cells from uh Advanced ad and 20,000 cells from non-ad individuals what can you use you can basically look at the distribution of values for those celles versus those celles everybody with me here seems like a reasonable
approach problem is that this assumes That every cell was sampled in isolation when in fact what's happening is that you don't have just a thousand cells or 20,000 cells from control individuals no we have 20,000 cells that came from 24 control individuals so the sampling process from which you obtain those cells is actually a little more complex it's a hierarchical process first you sample from a distribution of non-alzheimer's individuals a person and then you sample cells from that person And of course not every non-alzheimer's individual is the same and not every Alzheimer's individual is the
same so you don't have a gradient of cells you have 20 samples of cells everybody from here so now as you start thinking about differentially expressed genes for bulk data it's super simple because you have one measurement per group with single cell data it's more Complex because you have a thousand measurements for each of 20 individuals rather than 20,000 independent measurements everybody with me here so there's three categories of differentially expressed Gene analysis methodologies one is let's treat every cell in isolation and that one has a problem of you think that you have more samples
than you do because you didn't sample 20,000 cells you Sampled you know 20 groups of a thousand cells okay so then you're overestimating the confidence of your result because your variance is artificially lower because you've only truly sampled 20 people everybody with me here so that's one approach the cell centered approach at the other side you can basically say oh things were so wonderful when we're doing bulk analysis so I'm going to use the same statistics I'm going to take all thousand cells from each of my 20 People average them and now that's great I
have one measurement for that person okay this is perfectly statistically sound is it using the data to its full capacity no no it's averaging that variability and that variability is important because if I take a thousand cells and I see that they all have exactly the same value and then for those you know 20 Alzheimer's individuals my thousand CS have exactly the same value the Statistical difference between those is enormous but I've lost that because I've averaged them if by contrast those 20 samples each of a thousand sales have enormous variance and I look at
the you know control individuals and they also have enormous variance and yes their means are off by one but their variances are 20 each I really don't have any power everybody me here so basically this Approach of treating every sample as if it was bulk is you know very appealing very simple but it throws away a lot of the statistical power you have and in the middle is a mixture model that basically says I have a hierarchical process where in the first level of the hierarchy I'm sampling individuals and in the second level of the
hierarchy I'm sampling sales from those individuals and those methods are much more powerful statistically but they're Also much more complex a they take more time B they make more assumptions about the sampling process and those assumptions can in fact be sometimes leading you to the wrong result if your methods are not sound is everybody with me here so again there's cell-centric approach there's suitable approach where you're basically pseudo bulking you're basically averaging all of the self from the same person and then there's this Mixture model hierarchical type approach everybody with me here who feels that
they learning stuff yes awesome okay so then you can do these types of analysis and then say is there a difference in the distribution of values that are coming from say Alzheimer's or controlled individuals and let's look at some of the biological findings one of the things that we found is that if you look at differential Express gen means Between early and control in these two groups or between late and control then what you basically see is that in late stage Alzheimer's there's a lot more changes and many of them are shared between the cell
types whereas in early Alzheimer's you basically see much more cell type specific alterations is everybody with me here so that's one of the things that you can find out you can also ask how does sex biological sex play through the equation and what you See is that for example for non-alzheimer's individuals you find thousands of differences between excitatory neurons from male versus female individuals and you can do the same thing for Alzheimer's individuals and you find that the Alzheimer's cases versus controls sorry the Alzheimer's males versus females have many many more differences in their expression
patterns so that basically says that maybe we Shouldn't be thinking of just one uniform sample of male and female individual maybe we should be thinking of sex as a biological variable and sort of how that is affecting similar you can ask about the age of those individuals you can basically say oh well maybe some of these individuals were you know 99 years old and others were 89 years old and maybe expression changes with age anyway and maybe some of the differen that I'm finding are basically due to The age differences rather than the Alzheimer's case
control differences is everybody with me here again I'm not here to tell you all the answers I'm here to tell you how to think about all of these questions sounds good again you can do this with one brain region with prefrontal cortex but then again with when you look at how Alzheimer's progresses what you see is that there's a stereotypical progression it starts with something much closer to Your spinal cord at the at the base of the brain which is basically the locus celus and that's sort of the the place that start showing the differences
first then the second place is the Endo rinal cortex and from there you start seeing Chang in the hippocampus and then you start seeing changes in other subcortical regions and eventually start seeing changes in the cortical region so we basically said okay can we now study the differences That are happening in different regions of the brain between case and controlled individuals so we did exactly that we basically said can I look at the ento rinal cortex can I then look at the hippocampus the thalamus the mamary body and then some of the cortical regions the
neocortical regions the angular gyus the metal cortex and the prefrontal cortex everybody with me here what's the first thing that happens the first thing that Happens is that we go from having you know one class if you wish of exory neurons uh you know spread into different layers from that to col G there's many many different types of excitatory and if you look at the regions of the brain from which they're coming you basically see that they're actually showing dramatic differences between these different brain regions that some brain regions are showing only those Classes of
exelator neurons and then others are showing you know much more diversity the second thing you're noticing is that it's not just excitatory neurons and inhibitory neurons That Vary between different brain regions but it's also asites on dendrites and so so forth basically here I'm I'm toering those cells by the brain region and you can see here that there's a class of asides that specifically comes from you know different brain Regions than the other ones okay so that's the first thing the second is okay we now need to start annotating those additional classes of cell types
and we basically now have 30 excitatory neurons which I'm showing here and 23 inhibitory neurons and for every one of those you want to know what are the distinguishing features of those neurons what are the marker genes if you wish and then you're finding again dramatic variability between regions and also by Having many many more cells we're now talking about not 84,000 cells anymore but 1.6 million cells and of course technology has also improved from uh 2019 where we had the previous study and 2024 where we just published this study you basically and and this
just came out in nature um few weeks ago so you can also start sorting the cells based on how Alzheimer's like they are and you can start painting a progression of cells over time and that's one of the Things uh that you can do we can also do a lot of spatial analys I'm going to skip that for now and you can also say okay great I can study individual genes for how they're changing expression and then I'm doing 20,000 independent tests for every one of those genes about what changes in Alzheimer's disease for any
one of those genes I might not be very confident but if I Start seeing multiple genes in the same pathway then I might start gaining some confidence is everybody with me here but what if instead of analyzing the data one gene at a time and after the fact asking hey are those multiple independently measured genes changing in the same pathway What If instead I can analyze my data into groups what if I could look for this giant Matrix of genes by cells and then cluster that Matrix Decompose that Matrix using singular value decomposition and I
can basically say okay can I think about all of these single cells and all of these genes as being sampled from a lower dimensional space where instead of having every Gene acting in isolation I can think of igen genes or modules of genes that are acting together is everybody with me here so then I can basically say great can I group all of these genes together Into these modules and then start asking about first of all what are the genes that were grouped together into modules and I should say that your ta here Benjamin James
who's uh very busily in preparing and simplifying the P set that turned out to be a little too complicated um was in fact one of the leads for this analysis along with Carlos bch who um is now a postto uh over at the harbard medical school okay so basically what Benjamin and Carlos did is that they studied what are these modules actually meaning and uh they they you know you can you can take these Matrix and look at some just clusters or you can take these Matrix and start asking what is the correlation between every
pair of genes and the strength of that correlation between every pair of genes can give you modules of genes it gives you a network of genes and therefore now you can visualize every single Gene not Just if it's in a clust or not but where it is connected in that network of Gene Gene correlations if you wish is everybody with me here and you can use that to now start studying how mod of genes are changing in a coordinated fashion across different cell types and in relation with different pathological signatures associated with Alzheimer's disease you
can say what are the modules that are changing with cognitive impairment or Those that change with diffused plaques or neuritic plaques or neurop fiary Tangles or a combined Nia rean score for Alzheimer's pathology yes I have a question why are we doing this across clusters ofes and not just individual like what's the benefit of doing this in a cluster the benefit of doing it in cluster is that you have a more robust measurement because when you're measuring individual genes in individual cells the sample is just too small if One gene is expressed at four molecules
per cell and you're measuring individual cells the measurements themselves are going to be very noisy If instead you say well I'm not just looking at one RNA molecule but I'm looking at a dozen RNA molecules that are coar then it you more robustness does that make sense with P or any oh with bulk so the first thing that you lose in bulk is the cell types so basically instead of Saying oh it's this particular subass of microa then you'd be like how is this Gene changing between Alzheimer's and controls across all of the cell types
average together now with Alzheimer's you're losing neurons so it will look like oh wow all of these genes are decreasing in expression but those are the genes that are expressed in neurons what's happening is that you're sampling let's say 40 neurons per Experiment in this person and in in sort of let's say 4,000 neurons in a control individual but only 2,000 neurons in a Alzheimer's individual because they've actually lost those cells so then it will look like you have thousands of genes that are decreasing in expression does that make sense good so basically what single
cell analysis allows you to do is basically say what are the expression changes that are Happening within every one of those buckets of different cell types that's the that's the first level and that you can do with even suit analysis you can basically take all of the cells dissociate them measure 10,000 cells per person and simply condense that into seven measurements as if you had sorted exavator neurs you had sorted inhibitory neurs you had sorted aside sorted you know microa short vascular cells and then you basically did a bulk experiment In each one of those
that's fine too and you can do that and that's sort of the pseudo analysis that we're describing there make sense great these are all fantastic questions so keep them coming any other questions where's your answer doesn't need to be fantastic try to make the answer fantastic iist are clusters with different methods how actually yeah yeah yeah that that's a great question so basically we've now gone From Individual genes that I could just name H this Gene to modules like you know module 13 what does that mean to you and you know if I run the
analysis again will there be a module 13 again will that module 13 be slightly different the next time around so this is a fantastic question and again there's a lot of thought about exact reproducibility of the results my take on this is that exact reproducibility is almost a red hering And the reason for that is that if I were to do the experiment again and I would have sampled slightly different individuals what I care more about is the reproducibility across not just hey can I run it on a different computer but can I run it
with a different set of individuals or a different study or different cohort and so so forth and you can ask the question of robustness to some of those decisions for example if I subsample cells from the same people or If I subsample individuals and take all of the from those people or if I if I do both if I randomly subsample individuals and a randomly subsample cells or if I split the cells into two groups I basically artificially pretend that I have a replic experiment I basically take half the cells from this person and half
the cells from the same person but different halves and then I pretend that I have a replicate is that a true replicate the answer is absolutely not Because in carrying out the experiment I caught a particular region of that brain of that person of that donor and that region might be slightly different from the region that I did for the next experiment so what's extraordinary with these cohort is that there are two teams that carried out a very similar analysis Phi the Jagger a close friend and collaborator who basically was initially at the broad and
Has now gone off to Colombia who basically profiled 200 of these individuals separately and they did two separate replicates of the same you know for each individual for the same 200 people so we basically have something like 430 individuals for our cohort and something like I don't know another 400 individuals from their cohort 200 of which are the same so we now have three replicates two biological replicates From the same research group and one third replicate from our group for the same sample an awesome final project would be to say Hey how do these relate
to each other we've gone and parsed all of the data and prepared all of this but no one in my group is working on this so so hey and in fact as we start talking about projects tomorrow we have tons of data that is even unpublished so if if any of you guys are interested in sort of either diving into some of these Massive data sets or taking on completely unpublished data sets that are coming out of the oven you know in a couple of days you you can and um and again like the the
beauty of teaching this course is that this is not like 1906 physics this is like 2024 computation biology and this field is changing dramatically you know by the you know weak so so um anyway great question how do this change between replicates how do this change if I use a Slightly different version of the software will the results change yes yes they will change will that matter we'll find out right like that's what this is all about and um I'm so excited about you know seeing how many of these results will hold a test of
time but if you wait until you've done the same experiment seven times for publishing it as if we were waiting to sort of until the technology is fully mature to sequence the human genome I Don't know we'd still be in the Stone Age wondering if we should be using fire or not because fire can be dangerous right so so basically I feel that um you know time will tell you know we we publish a lot of hypothesis out there some of these hypothesis are replicated independently by other teams when you compare the field of Jagger
papers and the Ty Kelly papers you basically see that you know a lot of the similar pathways are in fact changed and that Gives us some confidence and you know the further we go into science the more confidence we'll have for some hypothesis and then some other hypothesis will realize we were wrong and then we'll just you know adjust change course and move on does that make sense anyway awesome questions and I'm totally enjoying answering these questions as you can tell so basically keep them coming raise your Hands doesn't need to be oh I don't
understand X it could be like hey I'm wondering about why who feels they're learning a lot raise your hands yes awesome good and who's excited about this whole field it's amazing right like like so much stuff is happening all right in the series of analysis that you guys can do for your projects yes of course you can study modules you can study um spatial uh transcriptomics you can study Epigenomics here's a very cool and very like crazy kind of analysis we basically said what is the correlation in the way that different cell types are changing
between individuals across brain regions and what we found is that for example the subiculum in the ental cortex was in fact you know uh highly correlated with the uh corresponding you know First Column here which is the um you know AGL you know whatever neurons Okay so basically what that teaches us is how the U different regions of say the hippocampus and the ental cortex are co- varing across individuals why is that exciting because it allows you to now say that oh across my 24 individuals this particular subass class of neurons was covariant with that
other subass of neurons and from again Decades of extraordinary Neuroscience we know that these regions are actually connected to Each other that they're projecting into each other so when you start thinking about how is Alzheimer's progressing in this typical pattern from the you know um Locus cillus to the Ral cortex to the hyppocampus ETC one of the hypothesis that pathology progresses either because of some physical damage or because of the way that the signals are altered and the neurons rely on those signals to basically stay alive and well So basically maybe the regions that are
signaling to each other are correlated in their damage because of one of these possibilities and that you know that that's another Super exciting analysis that you can do you can also start asking about how to group individuals and how to predict individuals based on their transcriptional signatures so this this is a different study in uh schizophrenia that was just published in science a Couple of months ago and what we basically did there is that we asked for control individuals and for schizophrenia cases can we predict whether a person is schizophrenia or control just based on
their expression patterns and what's really cool is that yes it did work for for many people that's yeah that that that's cool but what's even cooler is that it didn't work for other people and for this particular per Person control number seven the algorithm said wait that person looks very much like a schizophrenia person but this was a control so what's happening is that person in fact the case could that person have been misdiagnosed are our methods wrong are our signatures invalid well it turns out that this was the father of a son that was
actually diagnosed with Schizophrenia could it be that there's expression patterns that led to something inherited that basically was you know again schizophrenia is one a thousand so the chance that that person's sum is schizophrenic is actually quite small if it was completely independent so maybe there's something going on here conversely we found some schizophrenia cases that were just completely controll like in their expression patterns could It be that there are a different pathway that leads to schizophrenia like symptoms but it's actually not schizophrenia we now have 430 individuals with post morm single cell data from
Alzheimer's disease one of the projects could be hey I want to study those individuals that look normal but were diagnosed and see if there's something different between them I can basically look for either misdiagnosis or overdiagnoses or different classes of Al of Alzheimer's and so so forth so again there's you know an entire universe of data that you can do another analysis was looking at the Upstream Regulators that are Upstream of all of these genes that are differentially uh changing and then starting to predict what are you know what's common between these Regulators you can
also start studying uh you know genetic effects and so on so forth one of the directions and and you Know we've done this for pathogens we've done this for psychosis we've done this for ALS in front of temporal dimensia this paper was just published in cell just a few weeks ago um you know we're finding a lot of these Pathways that are changing uh Huntington's disease uh you know this was again published in Cell last year exactly year ago and this is looking at different subclasses of microa this is looking at vasculature this was published
in nature a year and A half ago um basically looking at the uh differences in expression in the vascular cells so vascular cells are are 0.3% of the data that we have and in the 0.3% of the data you can basically find a great of gene expression patterns between the vascular cells that are sitting on the venal versus the arterial part of the vascul and then seeing how these change in Alzheimer's uh we just published in Nature Neuroscience a little bit later And this was also published in cell about a year ago this is looking
at single cell accessibility uh and uh epigenomic differences in Alzheimer's this is um another type of that you can do which is basically asking can we infer from the same experiments the number of mutations that are there between the reference DNA and the sequenced RNA those mutations could Correspond to somatic changes that are at the DNA level or they could correspond to damage that happens at the RNA after being expressed but what's really interesting is that if you look look at Alzheimer's versus control individuals those that actually have dementia are basically showing more mutational burden
if you wish at the RNA level than the individuals that don't have diagnosis of dementia and that burden is happening very specifically in A subset of cells that we call the identified cells or Cent cells so basically you can actually use the RNA sequence to because sequencing to infer something about somatic mutations either at the DNA level or at the RNA level what's really interesting is that those individuals that show more somatic mutations are also the ones that that are showing more epigenome erosion and that's basically when the entire Global epome is just like flattening
out Instead of having clear active regions and clear repressed regions you know it's sort of you know diffusing out okay so I want to step back and think a little bit about this diversity of analysis that you can do at the Single Cell level and basically start reflecting about basically you start with your Ro data you then have count matrices of genes by cells and then you can carry out Quality Control Data correction normalization feature Selection and a lot of that is going to be in your problem set one that's going to go out on
Tuesday and you can then visualize the corresponding cells by projecting into the lower dimensional SC space and then you can start annotating them and then you can cluster the cells together then you can annotate these clusters based on different names then you can figure out the trajectory analysis as I mentioned earlier by basically looking at the number of splic Versus unspliced reads for different trajectories and sort of seeing where in that trajectory something is you can infer of course differential expression you can study loss of neurons for example and composition differences and so so forth
so one last uh analysis type that I want to mention is that in addition to this differential expression analysis which basically says I've now grouped all of the cells that I call microa Separately from all of the cells that I call excitatory neurons or microglia subtype 12 for example what you can do is actually start studying individual cellular neighborhoods and Brace yourselves because this might blow your mind what does that mean that means that I'm not going to look at discrete cell types anymore I'm not just going to Simply cut here and say all of
that is excitatory layer five or six okay instead I'm going To say within asites I'm going to study the neighborhood of every cell the transcriptional neighborhood of every cell this is not spatial neighborhoods this is transcriptional neighborhoods this is not XY coordinates of a two-dimensional plane that I'm looking looking at the cell this is some lower dimensional projection of the transcriptional similarity between cells so the proximity of those cells reflects how close are the transcription Everybody with me here and now because we have 430 individuals you can basically look at the phenotypes of those individuals
and project them onto those cells right this is what we call Self projected phenotypes so we basically pro project ad versus non-ad on those cells what do you see some cells are colored blue and other cells are colored red some neighborhoods are color blue and others Neighbors are color red that basically means that within the cell types and the subtypes there are now additional infinite resolution subdivisions that are basically telling you that Alzheimer's cells are moving towards here and control cells are moving towards there everybody with me here why is that exciting because I can
now annotate that neighborhood as more control like and annotate that neighborhood as more Alzheimer's like And what's really cool is that I can take an individual and forget about their phenotype and then go back and ask does that person have their micral cells in more Alzheimer's like States or more controll like States and the same person might have more Alzheimer's like microa but more control like pood dender sites are you with me here so instead of having the phenotype be something that I Measure once and I'm done you now start thinking about phenotype at the
level of individual cells and and that allows you to now completely increase the resolution of your analysis because you can then go back and reannotate your your individuals by basically saying how Alzheimer's like is every person in their oo dender sites and that gives you a measure ofite pathology that actually correlates with Additional measurements that you can do at the cellular level for the same people so through the Single Cell analysis you can do an extraordinary diversity of different types of annotations and you can now go from the neighborhoods to paint every cell Alzheimer's like
or not and to paint every individual and the sky okay needless to say that I'm excited about this field I'm excited about where this is all heading and I'm Really excited to hear your ideas about final projects tomorrow so we're going to meet in this room on Friday at 3 pm for recitation as always but instead of going over a problem set or additional material what we're going to do is spend the entire time going over your project so if and and your ideas and sort of what you want to do okay so if you
have ideas for projects now is a good time to start thinking about them read ahead from the material we've only had two Lectures which is very difficult to think about projects but think about the things that you care about and a lot of you guys have already submitted really awesome and very cool ideas tomorrow we're going to form sub teams and then we're going to start thinking about the types of project we can do in different uh parts and we start thinking about data sets from papers from unpublished collaborations that you might have from data
that we have in our group from Recently published papers from our group or other groups and so so forth everybody's excited who has not yet submitted their first day survey raise your hands okay awesome who has nice awesome aome great so please submit if you haven't already just sent you an email with a link and then see you guys tomorrow







![Wolfram Physics Project: Working Session Tuesday, Aug. 4, 2020 [Empirical Physical Metamathematics]](https://img.youtube.com/vi/VlqerIL2kl8/maxresdefault.jpg)





![Wolfram Physics Project: Working Session Wednesday, Apr. 29, 2020 [Finding Black Hole Structures]](https://img.youtube.com/vi/SbDzqlOLIZA/maxresdefault.jpg)

![Hands-On Power BI Tutorial 📊 Beginner to Pro [Full Course] 2023 Edition⚡](https://img.youtube.com/vi/77jIzgvCIYY/maxresdefault.jpg)
