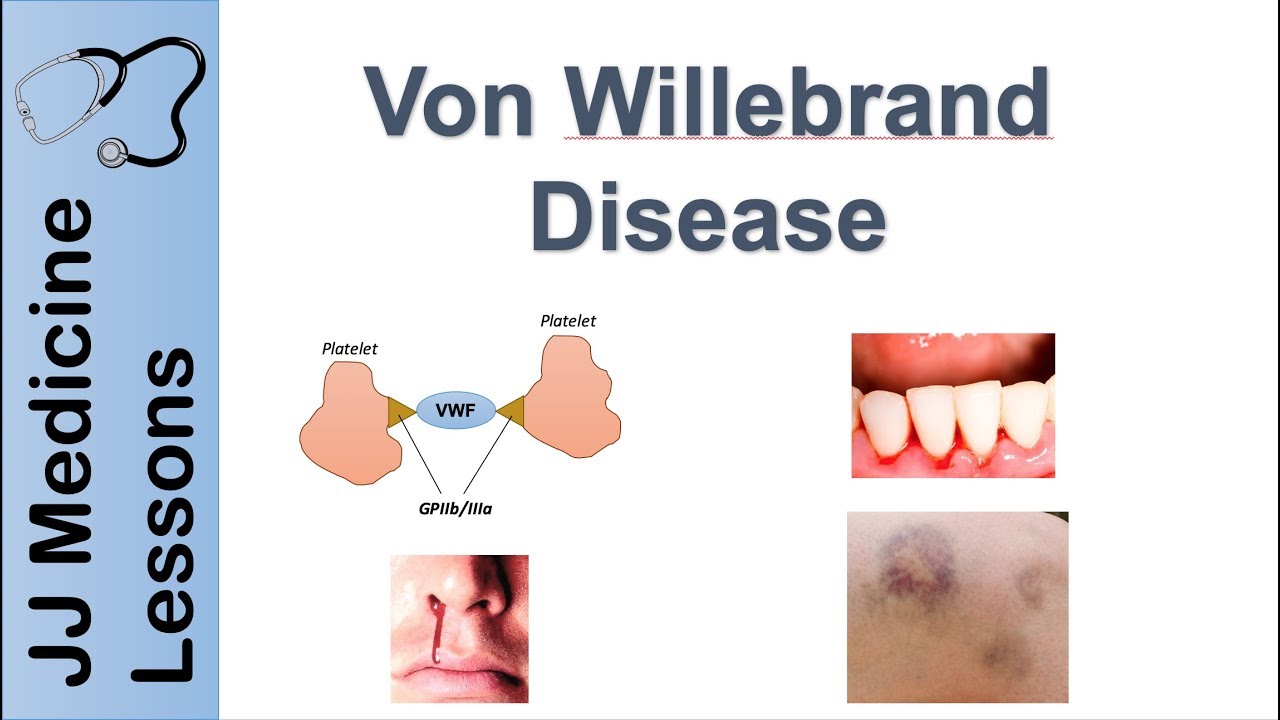
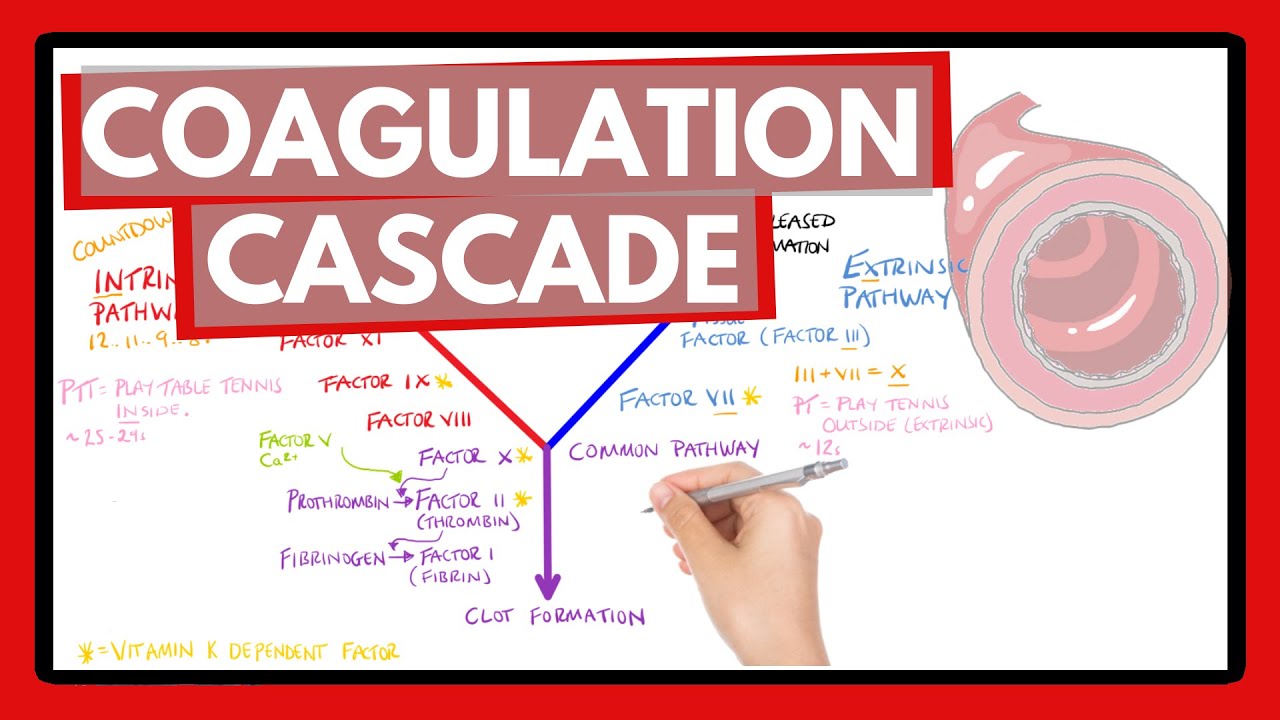
foreign hemophilia is a combination of the Greek words for blood and love a way of saying that people with hemophilia love to bleed or rather that it's hard to stop bleeding this is because the process called hemostasis literally meaning to stop the flow of blood is impaired normally after a cut and damage the endothelium or the inner lining of blood vessel walls there's an immediate vasoconstriction or narrowing of the blood vessel which limits the amount of blood flow after that some platelets adhere to the damaged vessel wall and become activated and then recruit additional platelets to form a plug the formation of the platelet plug is called primary hemostasis after that the coagulation Cascade is activated first off in the blood there's a set of clotting factors most of which are proteins synthesized by the liver and usually these are inactive and just floating around in the blood the coagulation Cascade starts when one of these proteins gets proteolytically cleaved this active protein then proteolytically Cleaves and activates the next clotting factor and so on this Cascade has a huge degree of amplification and takes only a few minutes from injury to clot formation the final step is activation of the protein fibrinogen to fibrin which deposits and polymerizes to form a measure on the platelets so these steps leading up to fibrin reinforcement of the platelet plug make up the process called secondary hemostasis and results in a hard clot at the site of the injury in most cases of Hemophilia there's a decrease in the amount or function of one or more clotting factors which makes secondary hemostasis less effective and allows more bleeding to happen now that coagulation Cascade can get started in two ways the first way is called the extrinsic pathway which starts when tissue Factor gets exposed by the injury of the endothelium tissue Factor turns inactive Factor 7 into activated Factor 7A a for active and then tissue Factor goes on to bind the newly formed Factor 7A to form a complex that turns Factor 10 into active Factor 10A Factor 10A with Factor 5A as a cofactor turns Factor 2 which is also called prothrombin into Factor 2A also called thrombin thrombin then turns Factor 1 or fibrinogen which is soluble into Factor 1A or fibrin which is insoluble and precipitates out of the blood at the site of the injury thrombin also turns Factor 13 into Factor 13A which cross-links with fiber and to form a stable clot the second way is called the intrinsic pathway and it starts when platelets near the blood vessel injury activate Factor 12 into factor 12a which then activates Factor 11 to factor 11a which then activates Factor 9 to factor 9A and Factor 9A along with Factor 8A work together to activate Factor 10 to factor 10A and from that point it follows the same fate as before so both the extrinsic and intrinsic Pathways basically Converge on a single final path called the common pathway believe it or not this is a somewhat simplified version of the coagulation Cascade but it has all the key parts needed to understand hemophilia now an insufficient concentration or decreased activity of any coagulation Factor can cause hemophilia except Factor 12 deficiency which is asymptomatic hemophilia usually refers to inherited deficiencies of coagulation factors which can be either quantitative or qualitative by far the most common of these are deficiencies of factor 8 which gives rise to factor 8A and is stabilized by another Factor called Von Willebrand factor and this deficiency is called hemophilia a or classic hemophilia another common deficiency is deficiency of factor 9 called hemophilia B which used to be called Christmas disease named after the patient who had it not the holiday now a mimic of Hemophilia a is Von Willebrand disease which is an inherited problem with primary hemostasis caused by a deficiency of Von Willebrand factor so in severe Von Willebrand deficiency factor 8 gets broken down faster and can become deficient too as opposed to inherited forms of Hemophilia one acquired cause of Hemophilia is liver failure since the liver synthesizes factors 1 2 5 7 8 9 10 11 and 13. also vitamin K deficiency can cause hemophilia since Vitamin K is needed by the liver to synthesize and release factors to 7 9 and 10. another cause is autoimmunity against a clotting factor and finally there's disseminated intravascular coagulation which consumes coagulation factors now the mutated genes in hemophilia a are called f8 and in hemophilia B they're called F9 and these are both on the X chromosome meaning both conditions are x-linked recessive so it usually affects men since they only have one X chromosome and therefore only one copy of the f8 and F9 genes women with one mutated Gene copy have a remaining healthy copy so they don't get hemophilia unless X chromosome inactivation turns off the normal copy in the majority of cells but generally women are carriers while men are symptomatic with the disease signs and symptoms of Hemophilia A and B are nearly clinically identical which makes sense since factors 8A and 9A work together in the coagulation Cascade to activate Factor 10.
both of these can cause easy bruising or echymosis as well as hematomas which are collections of blood outside the blood vessels that are often deep in the muscles prolonged bleeding after a cut or surgical procedure for example circumcision oozing after tooth extractions gastrointestinal bleeding hematuria which is blood in the urine severe nosebleeds and hemarthrosis or bleeding into joint spaces a dangerous complication is bleeding into the brain which can cause a stroke or increased intracranial pressure now the severity of the symptoms depends on the severity of the underlying mutation which determines the activity of the factor diagnosis of Hemophilia A and B usually starts with lab tests including a platelet count which is usually normal a Prothrombin time and a partial thromboplastin time the Prothrombin time tests extrinsic and common Pathways which means factors 7 10 5 2 which is prothrombin and one which is fibrinogen whereas the partial thromboplastin time test the intrinsic in common Pathways meaning factors 12 11 9 8 10 5 2 and 1. since factors 8 and 9 are part of the intrinsic pathway Prothrombin time is normal and partial thromboplastin time is prolonged in hemophilia A and B in order to confirm hemophilia ARB test to look at specific Factor activities and mutation testing of the genes encoding them can be done treatment for hemophilia A and B is usually done with injections of the missing or non-functional clotting Factor unfortunately if the patient has severe deficiency where intrinsic production of the factor is absent or very low these supplemental factors can be seen as foreign by the immune system which results in the production of antibodies that try to eliminate the injected clotting factors which are called inhibitors Inhibitors diminish the treatment's effectiveness over time and can also sometimes cause anaphylaxis which is a severe allergic reaction for hemophilia a desmopressin also called ddavp is helpful for patients with mild quantitative factor 8 deficiency desmopressin stimulates Von Willebrand factor released from endothelial cells which promotes the stabilization of the residual Factor 8.