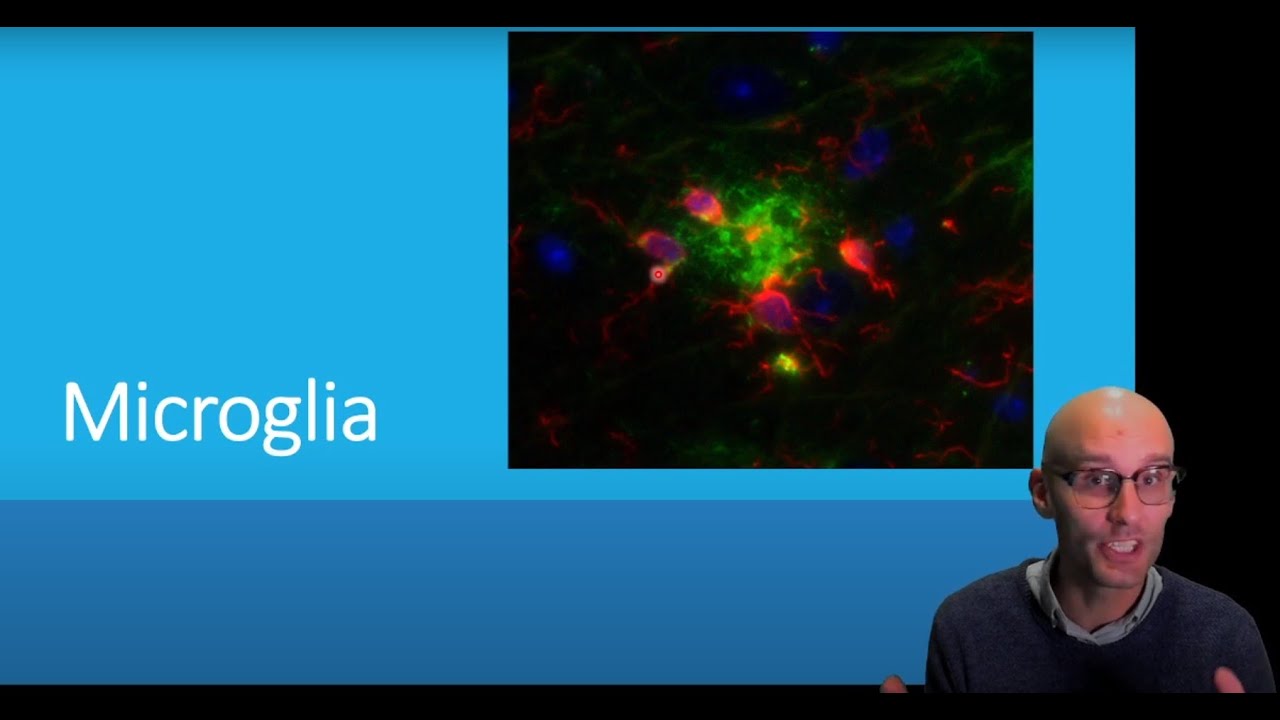
hey Tim Dog DJ Cordy and in the previous videos I've been discussing how neural inflammation contributes to Alzheimer's disease how the activation of microglare causes them to damage neurons directly and also recruit in damaging immune cells like neutrophils which also then go on to damage the neurons directly but how did the neutrophils know that the amyloid is there how do microglare detect amyloid and Alzheimer's disease now that's a super interesting question now another way to phrase that is what are the pattern recognition receptors for amyloid now we know microglia surveyor the extracellular environment using receptors
on the surface of their membrane or receptors within their phagosomes or even cytosolic receptors these are receptors that float around their cytosol to detect pathogens in there or damage Associated molecular patterns in the extracellular environment so the surveying the environment using these receptors that we call Pat and recognition receptors to say is there any diseases or is there any damage in the extracellular environment now these are called pen recognition receptors so which ones are these pen recognition receptors are involved in amyloid now this took us a long time this was a lot of research to
figure out this process and there was a lot of rabbit holes and a lot of dead ends in this research and that's because there are tons of pattern recognition receptors there are the toll like receptors the nod receptors the rip kinases for example we've also got the lectin receptors these detect carbohydrates like on fungus and then there are the inflammosomes for these are cytosolic receptors that respond to a number of different stimuli there are a lot of pain recognition receptors I don't want you to learn them all but which ones are involved in Alzheimer's disease
now I'm going to tell you basically what we've sort of concluded and then I'll jump into some of the research that how we discovered this okay so basically we think the primary signal is the signal here it requires three receptors on the surface of the membrane cd36 tall light receptor 4 until light receptor six and we think basically this is what's going to happen we've got the amyloid up there it's going to bind to cd36 ce36 is the binding and recognizing uh Motif that stands on your uh that sits on your outer membrane but it
doesn't have the sort of active secondary signaling molecules going on inside as a receptor it's just banned the cd36 now what we think happens next is that it forms a heterodimer now a dimer is two molecules two receptors Binding Together so that's tlr4 and tlr are six Binding Together and hetero means different so if it was tlr4 with tlr4 which does happen with LPS then that's a homodimer but with hitro it's um tlr4 and tll6 forming a heterodimer now when these two get together they activate a kinase cascade on the inside and it kind of
cascade is one enzyme phosphorylating another enzyme which phosphorylates another enzyme which phosphorylates the transcription factor which causes an upregulation of genes and it's how so many receptors work sometimes kinase cascade now this uses a famous intracellular part called Mighty 88 don't worry about that but that's just just so it pings in your ears if you hear it again so we get a mighty 88 which are the internal kinase on that receptor causing a phosphorylation Cascade and the end result is the phosphorylation of a protein that inhibits NF Kappa beta in your Kappa B sorry and
when that inhibitor is phosphorylated it releases in FKA B and allows it to go into the nucleus now when NF Kappa B goes into the nucleus it acts as a transcription Factor binding to promoter regions of the DNA and facilitating RNA polymerase to come in and cause the expression of a whole bunch of genes and these genes are inflammatory genes thorough inflammatory cytokine genes but they're also enzymes that produce reactive oxygen species which allow the microglare to attack whatever pathogen it thinks it's been exposed to and it's also enzymes that cause the production of inflammatory
signaling molecules like prostaglandins so the that's the primary pathway the amyloid interacts now let's have a look at the research there now basically they took microglare and exposed them to amyloid and then they genetically engineered the microglare to see how could we alter how could we block this response right so you could imagine maybe it's not one which is a an inflammatory pattern recognition receptor so genetically delete nod one and you notice that you don't change the response of the microglia to the amyloid therefore you know it's not nod one so you can go through
a whole bunch of these genes until you start to figure it out so this is what they figured out it's quite peculiar really if you knock out tlr4 from the microglare you block the response of the microglare to the amyloid now they also measured inflammatory cytokines but in this measurement they're measuring reactive oxygen species so when microglare are activated they spit out reactive oxygen species like peroxides like hydrogen peroxide in an attempt to kill the pathogen that they think that that detected so should a patent recognition receptor detect a pathogen they want to release those
reactive oxygen species like peroxide to kill the pathogen hydrogen peroxide you can imagine kills pathogens it kills everything but it kills pathogens so if you measure how much hydrogen peroxide another reactive oxygen species are coming off the microglare you're measuring how active they are really how inflamed and activated they are so they've got reactive oxygen species on the y-axis here the control microglia aren't producing any the amyloid treated ones are producing loads so here we can see lots of reaction species being detected or when they deleted the tall light receptor four they blocked the response
now an inferior researcher may go boom job done it's all through toll light receptor four but they actually wanted to check a whole bunch of other ones so they also notice when you delete tlr6 you block the whole response completely and again you might go okay job done but then they also showed that if you delete the cd36 received it on the cell surface you completely destroy the hot response so now you can see where that model came from if it was working through tlr4 and tlr6 you would expect a leaning one or the other
to maybe reduce the response by 50 percent but um or sorry tlr4 or tlr6 you would expect a leading one to maybe just reduce the response by 50 but deleting either of them to reduced it by a hundred percent kind of showing that you need them both together so forming a heterodimer you need those two receptors coming together in order to get the kinase activation and the production of those reactive oxygen species but then they showed cd36 was essential too and I think cd36 is doing The Binding of the amyloid and then bringing together tlr4
and tlr6 into a heterodimer and they showed that with a bunch of other experiments um and here it is here but this time with an inflammatory cytokine interleukin-1 now see if you delete LR4 or tlr6 you block interleukin-1 production and here they're blocking Mighty 88 now I mentioned mighty88 that internal domain That's essential for the kinase cascade so they're demonstrating that tlr4 and tlr6 are working through Mighty 88 as a kinase cascade and that's because tlr4 can sometimes work with other kinase Cascades such as triff and it depends on where the tlr4 is on the
cell membrane or within a phagosome and when tlr4 is facing into a phagosome then it tends to use trif as its internal kinase signaling molecule so this is saying the microgreen doesn't need to eat it it can just have it on itself surface and will get Mighty 88 kinase dependent kinase production of interleukin-1 beta so this is mRNA of interleukin-1 beta very cool experiment showing essential you need all three of those cell surface receptors to activate the kinase cascade and induce ir-1 production as well as reactive oxygen species so um ir1 is an example of
a cytokine that's an inflammatory signaling protein molecule but also there are other molecules involved in the inflammatory response and one example is prostaglandins so let me jump into those as well so prostaglandins are a small molecule there are produced by a Cox enzyme the digests the phospholipid membrane and it digests some of the phospholipids within the membrane to reduce this prostaglandin so cox2 is an enzyme that digests the membrane into an inflammatory singling molecule now an important point of that is because it came from a phospholipid it's actually a lipophilic molecule it doesn't like uh
it's it doesn't like water it likes lipids now what's unique about that compared to say a protein cytokine it means it doesn't really want to Flow In The Blood it was to diffuse out through all the membranes of the cells and through the fatty tissue and what that is great people is getting through barriers it'll get through any barrier so the blood-brain barrier for example blocks most water-soluble molecules from getting from the blood into the brain prostaglandins diffuse out and it creates that local response now prostaglandins also activate pain sensing neurons which is why prostaglandins
make it sore it's why often inflamed things are sore is one of the major reasons is these prostaglandins now this cox-2 enzyme is controlled by NF Kappa B so we know that if you activate tlr4 and tlr6 you activate NF Kappa B which causes the expression of this Cox II enzyme which digests the phospholipid membrane just a little bit of it it's not a major breakdown of the membrane it's just taking some of the components of the phospholipid membrane and turning them into prostaglandins and those prostaglandins then go on to act on prostaglandin receptors to
cause inflammation as well as pain um so a cox2 enzyme can be turned on by a damp or a pimp such as through tlr4 activation it can also be activated by cytokines so if a cell has an il1 receptor for example if you activate that il-1 receptor from using an il1 cytokine an interleukin-1 cytokine you'll get Cox expression as well um now it's slow and capable of prolonged signaling um and it's also quite local it doesn't flow around the blood it's more gently diffusing out through the membranes and that's because of the lipophilic nature of
it um now non-steroidal anti-inflammatory drugs these are drugs these are pain relieving anti-inflammatories that you buy off the shelf at the pharmacy such as aspirin and ibuprofen and Voltaren diclofenac methanamic acid there's a whole bunch of them but all the famous pain relieving drugs that you can buy over the counter basically are non-story went inflammatories and their main job is to inhibit the cox2 enzyme they also inhibit cox1 some of them and that's a complicated relationship affecting your stomach lining and blood clotting and all sorts of other things but the main goal is to inhibit
cox-2 which is that real enza-inducible enzyme that's associated with pain as well as inflammation and that's all through prostaglandins yeah and so it's it's primary regulator is any of Kappa B so the cox-2 enzyme is out here and if Kevin B causes the mounting of RNA polymerase all over the DNA and one of those genes is the cox2 gene that will now turn on in response to any of Kappa B activation okay next up we have cytokines um just to explain some of the differences um and they call they are again caused by pimps and
damps activating receptors or cytokines activating receptors often causing any capability activation and then that will cause gene expression now sometimes uh cytokines require approach translational modification so interleukin-1 and tnf Alpha for example aren't produced ready to go and they require sometimes a second signal and that second signal will cause the imagination they'll go from an inactive pro-molecule to an active mature molecule that's now released out of the cell but there are other cytokines that are just automatically released so they would have a signal peptide that would send them to the rough endoplasmic reticulum which would
then send them to be secreted straight away interleukin-1 does not have a signal peptide so it cannot get secreted straight away okay cool interference in that group too and a lot of cytokines not all but a lot of cytokines are regulated by NF Kappa B for example so again you tune in turn on in the fkb it'll go in and cause the expression of aisle sex il1 and tnf Alpha so they are proteins which means typically they are water soluble which typically means that they travel around the blood but they can sometimes struggle to cross
barriers so they're not like prostaglandins but to lipophilic they're more hydrophilic so they spend a lot of time in the blood or in the fluid compartment that means that they can signal long distances um so they are also slow and capable of prolonged signaling so there are faster ways to induce inflammation but the prostaglandins and cytokines are a little bit of a slower way to do it right so damps for example damage Associated molecular patterns are a much faster way to induce a local inflammatory response okay um a histamine is also incredibly quick anyway okay
so they're massively varied so cytokines can be anti-inflammatory and il-10 is a really good example that interlude containers and anti-inflammatory cytokine but these ones are mentioned here il-6 il1 and tnf Alpha are pro-inflammatory cytokines and chemokines I've mentioned this before are a subset of cytokines that attract upper concentration gradients so there are proteins that attract cells upper concentration gradient okay so why is tlr4 and tlr6 which are normally more associated with pathogens such as lipopolysaccharide from bacteria why are they activating in response to amyloid and that's a confusing thing because amyloid is a protein we've
produced it's not really a damage Associated molecular pen there's no damage uh amyloid doesn't necessarily correlate with damage it's just a cleavage product of a normal synaptic protein amyloid precursor protein so why are microglia receptors to taking this and it's an interesting question so I'm going to explain to you a technique that might help reveal it right so these are bacteria they've been streaked out on an agar plate and they've been stained with a stain called Congo red now Congo red binds to amyloid proteins and Conga red was first discovered as a bacterial stain to
stain amyloid proteins and amyloid-like proteins that are expressed on bacteria so we can see that these bacteria are testing are positive under the conga red stain showing that these bacteria have amyloid blight Pro proteins protruding from their cell wall now we can use that same Congo red stain to stain Alzheimer's disease so here this is an Alzheimer's brain and in red hair we have the plaques now the amyloid plaques are staining with a stain that we normally use for bacteria and this might be your first hint perhaps these amyloid oligomers and amyloid fibrils are molecularly
very similar in structure and properties to proteins that project from the cell wall of many pathogenic bacteria so perhaps it is an example of molecular confusion perhaps we our receptors that are imperfect they've just evolved to detect bacteria happen to also detect amyloid and amyloid fibrils and then when you think about this in terms of evolution right um Alzheimer's disease typically hits people at the age of 70 that is well after their reproductive period Well after their neutral nurturing of siblings period and for most of human history having a negative effect on a 70 or
75 year old survival such as Alzheimer's disease would not have a strong evolutionary pressure there's no longer not a lot of reason for these tear LR4 and tlr-6 receptors to evolve not to detect our amyloid especially when you consider one of the other threats which is bacterial infection they need to detect those bacteria because that will kill you as a baby that will kill you as a 10 year old that will kill you as a 20 year old right when you're it's very important from an evolutionary perspective so the evolutionary pressure is for us to
make mistakes it's better to be sensitive and not miss a bacteria and occasionally mistake a protein that's associated with old age is a pathogen than it is to there's not much of a selection pressure to avoid detecting our own amyloid that builds up in our later life right so our receptors are probably just hypersensitive to amyloid-like proteins and there's no pressure for them to figure out is not a good idea to respond to these amyloid plaques is if the pathogens Evolution has not prepared us for that okay so that's just a theory and but it's
a reasonable Theory um but it is just a theory about why our pen recognition receiversity designed to detect bacteria typically have evolved largely to detect bacteria are now detecting A non-bacterial protein that we produce that's possibly why okay so does amyloid look like a pathogen essentially however it might be a long video sorry guys uh so now these are the plaques right these are soluble so they're not floating around in the fluid so they're not available to sort of bind to receptors right and amyloid football is really going to struggle um when it's not sitting
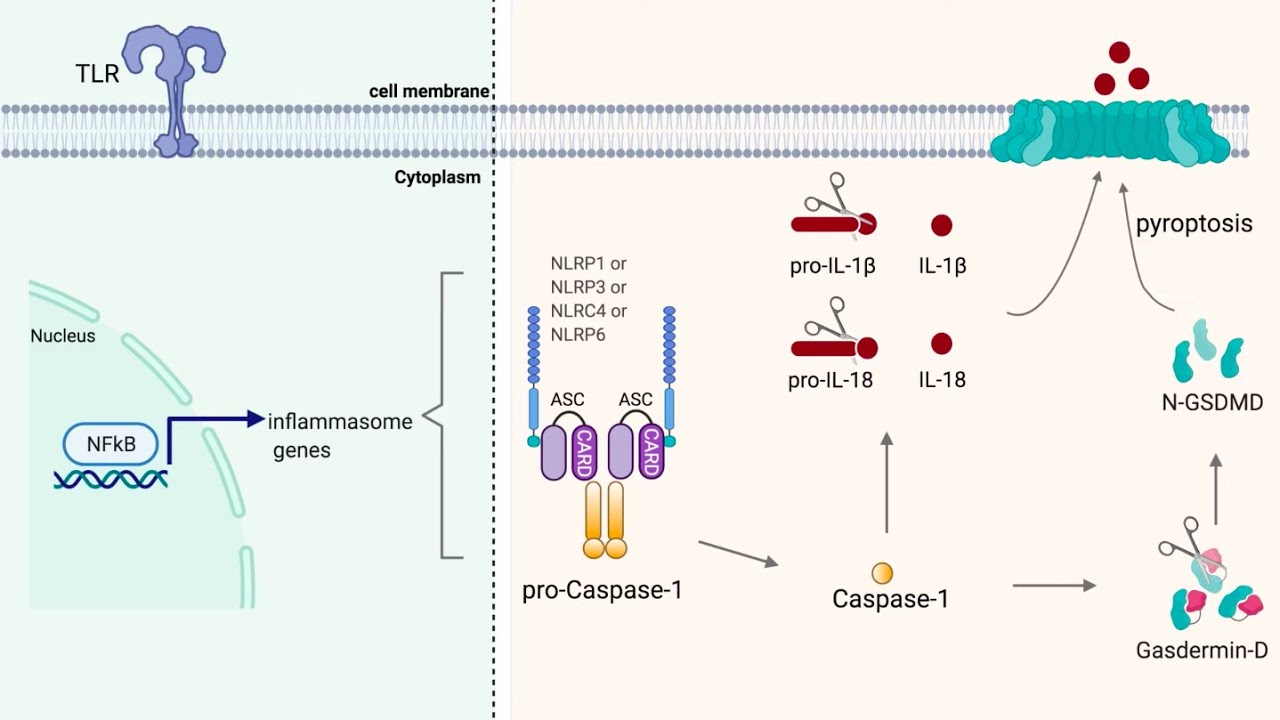
in solution to bind to a small protein receptor on the surface of the membrane also you know they're often as big or about half the size of a microglare so they're not going to be binding to a little receptor something else is going on there so how do them amyloid fibrils activate the immune response and that's when we come into some of my research this is this has been you know seven years of my life so far has been researching this topic and it's this topic of these proteins called inflammasomes in particular one called nlrp3
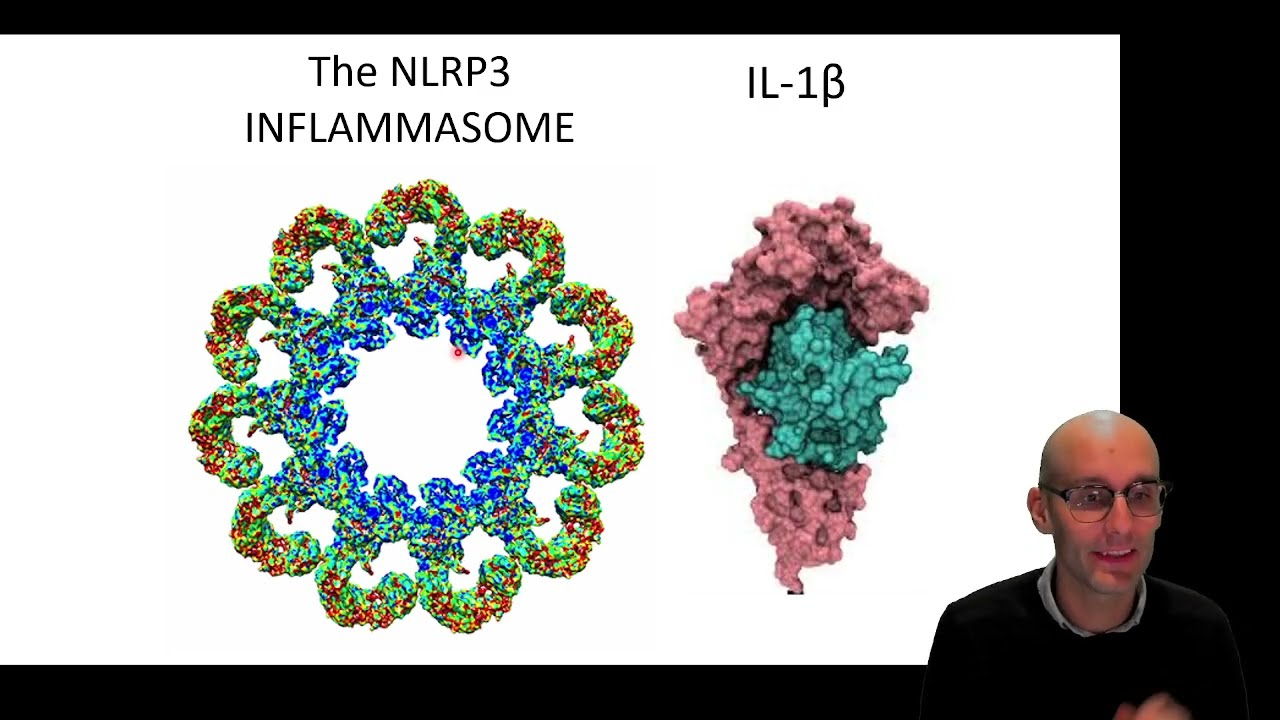
what it stands for it doesn't really matter a lot of genes don't really mean anything their names um it's not like receptive pyrene containing protein three okay but it doesn't matter it's the nlrp3 everyone knows it is in lrp3 and Alzheimer's disease so this is the nrp3 um inflammazone now each one of these just one molecule here is an nlrp3 Protein that's the receptor that's the patent recognition receptor it's an nlrp3 protein and it's oligomerized into the thing that we call an inflammasome and that's when it's activated it all sticks together in this ninja star-like
protein is gosh it's a beautiful molecule you should really all just research this look how good looking that is um and it forms this complex when in fact it now there are links to enlar P3 and Alzheimer's disease so let's jump into that now okay so it all ties into interleukin-1 beta I've talked about interleukin-1 beta as one of the most inflammatory cytokines we've got in our body well almost every video on this channel because this is what I research into Pokemon beta but the S the story of interligible media is quite complicated so here
we have our NF Kappa B and it's bound to the DNA it's mounted the RNA polymerase it's called the expression of the gene of interleukum beta and is now being produced however it's produced as an inactive form Pro interleukin-1 beta it's not active okay it's a large molecule that's not active essentially it's got a big protein stuck onto it to inactivate it it's like if you have a key and then you weld a big chunk of metal to that key that key now cannot fit into the lock and activate the lock so it can't activate
the receptor it's got a big useless chunk attached to it so we need to chop that off before it's now mature into Luke and One beta so we chop the chunker off and now the key will fit in the lock and open the receptor so we need to chop it off in order for it to be activated and indeed in order for it to be secreted now as char by an enzyme this enzyme is called caspase one this caspase one actually chops a whole bunch of proteins but one of the proteins that chops is pro
interleukin-1 beta and it converts it into the mature form by chopping the useless chunk of protein off called the pro domain care space one however is also produced in a negative form it's a pro enzyme a zymogen if you will and so it needs to be activated too that's when my baby the nlrp3 inflamasome comes in so it normally sits as a monomer in the cytosol there then it detects a disturbance in the cytosol something changes in the cytosol and that will activate that in lrp3 receptor it will then oligom rides into the ninja star
that's called the inflammasome and that will bring in an adapter molecule called ask and it actually brings in so many ask adapter molecules that it becomes as big as a mitochondria it's so huge it's just a protein complex but it's massive and that's why we call it an inflammosome because it looks like you know a lysosome you know it's as big as a mitochondria it's like two microns wide it's huge each of those S molecules can recruit a caspase and activate the caspase and it actually forces the two caspases it forces two caspases together to
activate themselves it's called Auto catalytic self-catalytic so the two caspases get brought together by the ask molecule and it activates and you end up with active caspase one which cleaves the pro ir1 into mature ir1 super super cool and this is called the inflammazone complex um and this is that post-translational modification I mentioned when I talked about cytokines so sometimes cytokines are produced in an inactive form they require some sort of post-translational modification like cleavage in order to become active and I you might ask why I had this two-stage thing well interleukin-1 beta is one
of the most inflammatory cytokines in your body if I inject it into you you will immediately get a temperature if I inject too much you'll probably die from leaky blood vessels from too much of an immune response too many activated neutrophils dropping blood pressure all their Jazz right so it's kind of a nuclear bomb and like a nuclear bomb it's good to have two keys that need to be turned in order to launch the nuclear bomb so the expression of interleukin-1 and the expression of vanilla P3 is one key and then the activation of in
lrp3 is another key in order to get the release of interleukin-1 beta so that's why I think there's those two barriers because you don't want to accidentally turn on your interleukin-1 system it's going nuclear when it comes to an innate immune system right but it gets more complicated and deliver One beta still can't get out of the cell really there are some subtle ways but for the most part interluka one beta mature struggles to get out of the cell so we need something to happen so we have you should all be familiar with apoptosis which
is program cell death the cells glib they pop out phosphatidylserine and they also send out signals to the uh microglia and the macrophages come eat me and they get in there's a necrosis which is uncontrolled cell death that's like a squashing or a popping by a toxin and it means that the cell's gone so dysfunctional that it can't die in a safe way and it just releases everything it's gross next we have pyroptosis now pyroptosis when an immune cell fills itself up with cytokines and then pops and when it pops it releases the cytokine a
huge amount of it and that's what happens with interlude beta it's called pyro because um interleukin-1 used to be called a pyrogen because does it cause your body temperature to go up it causes you to get a fever so it's called pyroptosis because it's a fire pyro you know pyromaniac it's to do with fire it's to do with temperature it's an inflammatory way to die pyroptosis super inflammatory it's a fiery way to go it's pretty cool so that's what happens with uh um uh that's what happened that's how interleukin-1 typically gets out of the cell
there are other ways but for the most part it happens through pyroptosis in the cell popping and releasing into Luke 1. so let me now go step by step on in lrp3 and amyloid because it's a bit more complicated than what I've explained even though it's super complicated what I've explained but I gotta tell you about this because this is amazing okay so here we have februles oligomers and monomers of amyloid right as I've already discussed oligomers and monomers have the ability to bind to the cd36 and to cause the heterodimer of tlr sex and
tlr4 so the monomers and oligomers are doing this they're activating these receptors these receptors once activated will then cause the inhibitory molecule of NF Kappa B to come off because it will get phosphorylated and stop hitting it inhibiting it it's called ikk so that will come off after being phosphorylated and now NF capital B will go in bind to the DNA and it will Mount RNA polymerase and it will cause the expression of lots of things including things like il-6 tnf Alpha but also Pro il1 beta Pro Cast Space 1 and nlrp3 ask was already
being expressed over there and ask is kind of constitutionally expressed in the cytosol so now we've got in lrp3 Pro il1 and procast phase one in the cytosol ready for their activation we need that second step now so next we have oligomers and februles binding to phagocytotic receptor complexes basically the membrane is not great at just taking stuff up when it often needs receptors on the surface there to help take up fibrils and oligomers and it will now bring it in as a figure lysosome but there's a problem especially with fibrils but also oligomers the
lysosome the phagol lysosome can struggle to digest what's inside it and actually the amyloid can end up bursting the lysosome so the license fegolysosomes can end up rupturing and when that happens digestive enzymes come out and we end up with a lot of problems so the phagol lysosome has now ruptured and what this can actually do is it can compromise your membranes it can activate receptors and it can also activate enzymes that create pores that form in your membrane and it can actually damage the membrane itself so now the membranes become leaky we end up
with potassium efflux and chloride efflux now both of these things are essential so potassium and chloride rush out of the cell because of receptors as well as pores being formed in the membrane and that potassium and chloride efflux causes the nlrp3 to oligomerize into its inflammosome so the potassium and chlorating flux has caused nlrp3 activation there once we get into lrp3 activation we give the caspase one activating and the cancer phase one Cleaves Pro interlium One beta into mature interleukin-1 beta however the material into liquid still can't really get out of the cell all by
itself it needs something else to happen so what happens is active caspase 1 goes on and it activates another molecule now this molecule is called gas doom and D now gas doom and D once activated can form a pore in the membrane so it's inactive it needs to be cut once it's cut it'll ligamerizes into a pore and they're poor right there can allow the mature interluka One beta out of the cell it can also kill the cell and that's pyroptosis so the the pores forming from the gastderman D causes pyroptosis which is very very
cool and I understand that a super complicated pathway but it's super important and it's so fascinating there's even a million more other things you could explain about it biology is kind of fractal you could zoom in on this one bit say here and it gets infinitely more complicated nlrp3 is bind to endolysosomes it's ubiquitinated it must be de ubiquitinated it must come off the figure oh it must have come off the endosomes and blah blah blah there's a million things there's a neck seven and there's another molecule any bet within here you can zoom in
on and it will be super complex that's why biology is fractal you zoom in it stays complex it's amazing it's a never-ending well of research which if you want to jump into it okay so this is the guess doom and D so here we have it and it's inactive it's got this orange chunk bound to it which deactivates it you cut you cut it and now it will insert itself into a membrane and will form these beautiful pores in the membrane and ir1 can now come out of it there's some evidence that the pore is
a little bit selective and it will mostly let ir1 out and not let some other things out but eventually enough pores can form in your membrane that the cell will pop and then everything comes out and that's called pyroptosis all right so how do we know enlrp3 is important in Alzheimer's disease well you might have seen this experimental Paradigm before it's very common you get an inner rp3 knockout Mouse this mouse has no nlrp3 receptor you cross it with an Alzheimer's mouth so it's an app PS1 it's got two familial mutations that we know cause
Alzheimer's disease one in the app protein one in the game of secretes and what happens is you cross those and you'll end up with after a few Generations wild type mice in lrp3 knockout mice Alzheimer's mice and Alzheimer's mice without nlrp3 right okay so now we've got these mice let's test them what happens in the Alzheimer's disease so we use the Morris water maze again so these mice have been trained to find this platform over five days then we remove the platform and see where the mice one now a mouse with a good memory goes
whoa I'm pretty sure so we're looking down upon the Maze and we've tracked the mouse the trail of the mouse and here's the hidden platform there and this is the water mates there so we've trained it to find this platform and it's just swimming around that platform it's like I know the Platformers here I know the platform is there but I can't find it because we've removed it but we know that this mouse has remembered where the platform is over the five days because I was looking for it where the platform should be this is
an album's Mouse I can't remember so it's just doing a general surge pressure search pattern it's like it is forgotten the previous five days of training it's got a poor memory it's forgotten all its training and it's just swimming round and round in circles it's got no idea where their platform is despite it's been trained for five days to find it here is an Alzheimer's Mouse the inner rp3 knocked out so it's not releasing the io1 in lrp3 is not being activated so it's not having that inflammatory response there's probably no neutrophils coming in there's
probably no microglial phagocytosis some neurons going on no release of reactive oxygen species none of that kind of stuff's going on and what we can see is uh the Mouse has a perfectly good memory so if you inhibit nlrp3 activation and Alzheimer's disease you delay perhaps prevent uh the amyloid and Juice cognitive deficits that we see in these animal models um and this is just the training data so this is the mice getting faster and faster and faster they're traveling less distance there's actually a measure of distance so they're traveling less distance to get the
platform these guys were actually trained for eight days and in black and blue we have wild type and in the rp3 knock Mis and green we have an Alzheimer's mouse that is also in lrp3 knockout and in red we have an Alzheimer's Mouse so in red the Alzheimer's mice pretty much don't get better throughout the whole eight days of training whereas in green the Alzheimer's might sort of inner Lupita milk out do learn over there eight days and get as good as the wild type mice uh they also measured the amount of mature interleukuma beta
in the brain which is a very hard thing to do so they're super impressive because you can see it's femto grams tiny tiny amounts there um are being released and we can see we've got mature R1 in the brains of Alzheimer's mice and no mature Isle one same levels as the wild type basically in The nlrp3 Knockout so we're blocking ir1 release and we're getting better cognition in these mice of the inner rp3 knockout this leads to the hypothesis of a paper that I'm about to break down as my paper which is the factors which
influence enlarp3 activity and i1 secretion will affect Alzheimer's disease progression I've got three big papers on this topic I've got two papers on can we inhibit nlrp3 as the therapeutic Target for Alzheimer's disease ones in animal models and one is in looking in human data which is very cool and over here is what about can we sensitize in lrp3 through environmental factors that accelerate Alzheimer's disease and I mentioned in this in the next video um I look at zinc deficiency because we know zinc deficiency sensitizes enlarp3 we also know it's one of the most common
malnutritions in the world so perhaps if you're zinc deficient you've synthesized in lrp3 you're going to have worse Alzheimer's disease and that's what I show in a paper and in that paper I use human data Mouse data and cell data I'm very proud of that paper too I'm not going to break that down though I'm going to focus on on inhibiting email rp3 as a therapeutic tag of Alzheimer's disease in the up and coming videos and that's the paper right there that's what we're going to break down it's using insides to inhibit nlrp3 to protect
it get Alzheimer's disease oh thanks team next video is on whistlebots