I'm W gong I'm assistant professor Northeastern uh and I have been at the shimi center for three years as a postto uh it's is my great pleasure to uh introduce Hillary fukin uh she's a assistant professor at har medical school and Associate member at the broad and uh assistant in investigation in the translational genetic unit at MGH today she will talk about her work exciting work on variant scoring and we are all Eager to hear what she has done in the space so with that without further Ado please take it away uh thanks very much
for that introduction um I'm really excited to uh be talking to you guys today thanks very much uh for the invitation um to speak at this uh at the Symposium um so I'm going to start with genomewide Association studies uh uh as many folks in this audience will be familiar with um genomewide Association Studies have Now identified tens of thousands of regions of the genome or genetic Loi that are assoc iated to common diseases and complex traits so here's an example of a now old gwos for schizophrenia where you can see genomic coordinates on the
x-axis each point is a variant the significance of Association is on the y- AIS um and by analyzing genotyping data from tens of thousands of cases and tens of thousands of controls researchers were able to identify over a hundred uh Gen genetic Loi at which genetic differences lead causally to differences in disease risk uh each one of of these green diamonds represent such a Locus um and I just want to pause there even though this is not um news at this point but just to re-emphasize the fact that we're able to make statements about causal
human biology that leads to disease is really exciting um uh but of course the big problem that the field faces now is we have all of these Genetic Loi but how do they lead to increased disease risk this is an enormous challenge for for um uh uh this field we have these really exciting promising little windows but for each one how are we going to figure out um the actual mechanisms by which these genetic differences lead to differences in disease risk um so today I'm going to talk about uh two pieces of this puzzle one
piece is how can we take a region of the genome uh and refine it into the Specific set of variants that are causally driving this difference in disease risk um and then what's a roote towards starting from these variants that we have and trying to push forward towards um the the next of understanding what they what they do um so uh to start by uh talking about identifying the specific variants um next slide uh oh is this a can someone in AV help me advance a slide there we go Oh back a couple now there
we go okay so uh my group a couple years ago performed fine mapping in three large scale biobanks before I describe the results I just want to take one moment since this is um an audience with a lot of machine learning folks to talk about the methodological piece of this um so I'm not going to say any details but I'll just uh just imagine that you have to do variable selection you have tens of thousands of features the features are Highly correlated with each other you have hundreds of thousands of samples um and the kicker
is we really care about accurate quantification of uncertainty you can't do lasso you mcmc is way too slow for this uh size data very variational inference is also not accurate enough um so there's been a lot of really creative work figuring out um how to do uh work uh you know approximate these posteriors in ways that's accurate enough to be useful for What we're trying to do but fast enough that we can then apply it to you know our 108 uh lowside that we've identified it in our gws so uh and my work has done
methods development um work in this space my group has done methods development work in this space um a lot of what we've done has focused on um robustness to model Miss specification um and how can make sure that the results that we get at the end of the day are as reliable as possible so we Did find mapping next slide yes um in three biobanks uh so each of these biobanks has over 100,000 um individuals we're looking at tens of different phenotypes um by doing associations on all of these phenotypes at this scale uh there's
16,000 Losi toine map um and uh this was approximately order of magnitude a million compute hours to um narrow each of these lowside down into the list of the most likely causal variants um this was led oh thank You excellent thanks um so this was led by PhD students masak Kanai and Jacob ulish uh and co-supervised by Mark Daly and it was an enormous effort which um is described in more detail at that link um we identified thousands of likely causal variants uh and while there's some caveats around the um uh posterior prob ility of
causality and and how calibrated uh we think these numbers really are this still represents a remarkable Trove of variants that are Likely to have uh causal impacts on disease biology to characterize these variants uh we started by breaking the whole genome down up into five basic categories um exonic utr promoter enhancer and other and you can see genome wide most everything is other but as you start to uh break down the um categorizations uh oh of next slide this one doesn't seem to be working either um of these different variants there we go then you
can see That uh you see really large enrichments in these different categories with the most highly confident variants um really strongly enriched in the um coding uh coding category especially so now um let's pause for a second and think about how cool it is that we've got uh fine mapped missense variants so because of this really large scale computation that we did and because of the methods development work that's been done in This field we can Now list 1,391 unique missense variants that are causally contributing to common diseases and complex traits and so if we
back up to our original question of we've got all of these Loi how are we going to translate these Loi into biological uh insights now we've taken a really important step which is identified the individual ual variants and for these missense variants we can even say uh what the next step is these are Mutations that cause one amino acid to substitute for another amino acid at a particular position of a particular protein um and that is really um uh you know a big step forward from analyzing DNA mutations but now we have to figure out
what these um amino acid substitutions do uh and amino acid substitutions of course they can do a ton of things on the lowest level you've got a different set of atoms in different places at this point in the Protein and that means they're going to exert different forces on the other atoms that come near them uh oh but that's only interesting because it means that the protein is going to be expressed in different quantities or different places or it won't be able to adopt the right confirmations in response to the right stimuli at the right
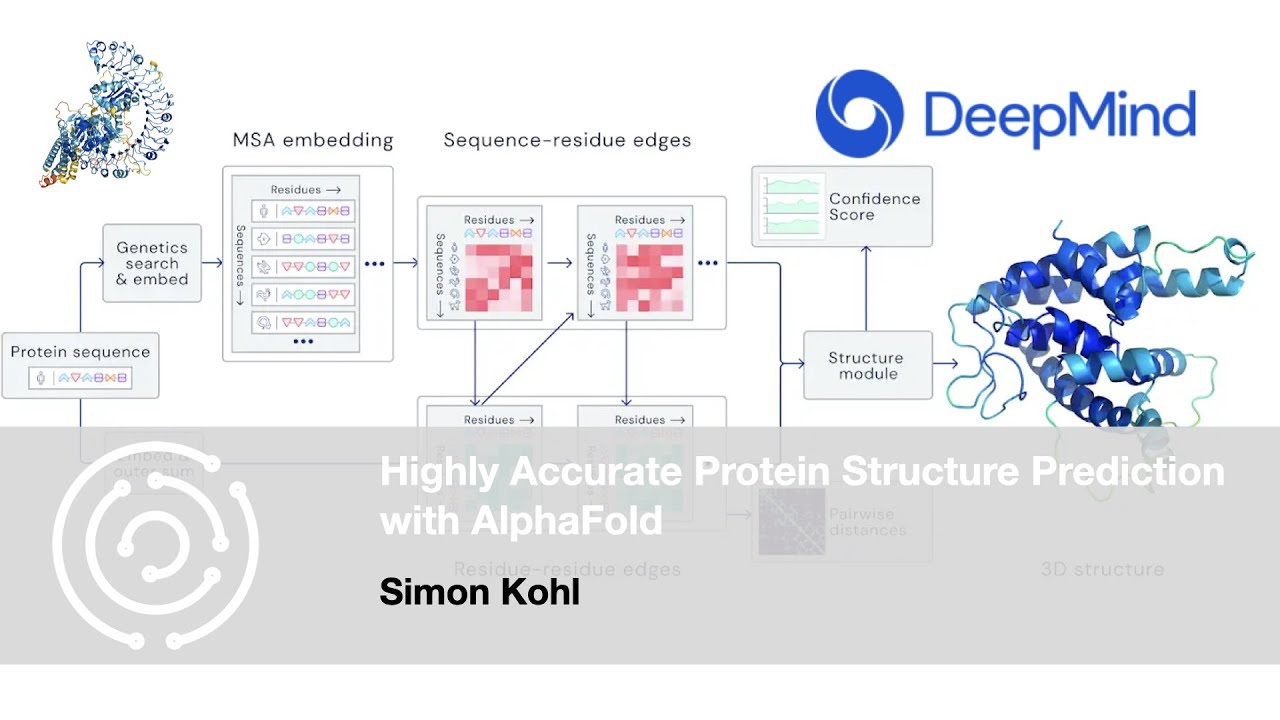
times so this is a hugely complex multi-dimensional problem that we still are fac faced with uh and this is where The AI comes in so uh protein modeling uh as as Lucy um um uh talked about in the first talk has made enormous strides um and this is a really exciting field we now have seen a lot of uh leaps forward and prediction of a lot of different uh for example structure from sequence of proteins a lot of these fundamental questions um so how about AI for understanding the effect of missense variance um predicting the
effect of missense variance is an extremely field So the atlas a variant effects Alliance uh conveniently keeps a table where they're uh keeping track of all of the methods to predict uh the effects of of variance and it starts from classical methods like polyphen or sift but if you scroll down uh then you can see it goes all the way down to 94 uh methods as of pretty recently there's an extraordinary amount of research in this space um and a lot of these methods are um getting a lot of attention because they're solving A really
important problem um uh and so the uh so this is a field in which there's been a ton of methods development and the methods development has been extremely sophisticated also it's uh reading these papers you can see a very wide range of tools uh being brought to bear on this really important problem so folks are using uh message passing neural Nets on on uh graph derived from protein structure uh people are using extremely large language Models trained on protein sequence databases which I'm excited to hear more about in a future talk here uh variational autoencoders
are being trained on multiple sequence alignments the embeddings from the protein language models are being fed into population genetics models the protein language models are being combined with the variational autoencoders and then calibrated using gausian process priors alphafold fine-tuned on population Genetics data with an auxiliary structure loss um and you know convolutional neural Nets on voxelized protein structures supervised on population genetic data with auxiliary losses from both A variation Auto encoder and a protein language model and a masked structure objective like this field has just gone uh has has really seen a whole lot of
sophisticated methods development um so with all of the you know brain power and Computational resources that have been brought to bear on this problem we were reading these papers and wondering so which methods are are best and how good are they um and that is a lot less clear than the fact that there is really cool methods development going on here so so just to give one um uh kind of set of anecdotes from a series of papers one task that you might hope a pathogenicity predictor would be good at is differentiating between denovo mutations
Found in cases versus denovo mutations found in controls for severe disease um and a lot of these papers are doing benchmarking on exactly this task but they're doing it in very different ways and getting very different answers so for example in June 2023 uh we were seeing uh benchmarking on Den noos uh using the man Whitney U test uh P value uh shortly afterwards uh different set of data different uh different metric now we're looking at Aur um and now here it's again denovas for severe disease but again the classification task is a little bit
different um and we're using Precision recall here different data different classification task we're still talking about denovos for severe disease but we're using a fourth evaluation metric and all of these papers are coming out so quickly also that they're not um the methods aren't all being compared to each other and so it's really difficult To kind of read through this uh material and understand where are we at as a field right now um so we came to this thinking it's time for some independent benchmarking using human genetics data using the metrics that we know are
most informative about the outcomes that we care about the most um and one big inspiration for this work is that um in the Deep mutational scanning field there's been some really uh great efforts to systematize the benchmarking And then to have independent groups come and Benchmark a whole bunch of these different methods um but for human genetics things are still pretty scattered um so we wanted to ask three main questions with this work uh first is just a metrics question what should we be using as our metrics to evaluate these methods um and then also
to expand past there's one dimension on which methods can be better or worse at and instead say uh there's a wide range of Even within pathogenicity predictions there's a wide range of possible pathogen to City tasks so how are these methods performing um at different parts of the selection Spectrum um and also how does performance differ for within Gene tasks versus across Gene tasks which are two very different tasks um so Conrad keski uh uh and uh Sophie parsa and Jeremy gz in his lab um uh together with masaai whose name got cut off the
slide apologies Masa um together with Several other collaborators uh and I set out to do this independent benchmarking um and we are calling our framework genetics gym inspired by a protein gy from the marks lab um and we did four main types of um benchmarking to begin with so all the way on the heaviest selection end of things we are returning to denovo variants for severe disease um and then now we're looking at rare variants for Neuropsychiatric disease rare and low frequency variants for Common diseases and complex traits and then common variants for common diseases
and complex traits which are these fine mapped variants that I described in the first part of the talk um and so here are a subset of our results that we um uh uh that we found using this benchmarking on a subset of the data and I think that there are a few main takeaways here one of them is that the methods that are doing really well in the heavy selection regime are Different than the methods that are doing really well in the lighter selection regime and this isn't that surprising these are very different tasks um
and and selection is doing very different things in these different places and a lot of these methods are trained with a lot of evolutionary data um and so it makes sense that that we would see really different um tasks but I hope that or different performance um but I hope that what this does is it Allows the field to start to move away from saying we have the best pathogenicity predictor to saying here's the particular area that we're focusing on here's the area that we now are doing well at um and that we're going to
be able to um see papers that instead of saying we're the best at everything say like this is specifically what we're good at and here's our relative performance at these other tasks um so When you're doing pathogenicity prediction and you're trying to differentiate a set of disease variants from a set of control variants to identify those variants there's two pieces identifying the Gene and then identifying the variant within the Gene and uh we wanted to tease apart uh those two different components um because looking at this it's really unclear to what extent for example are
popiv and Misfit um methods that are doing really Well because they can find the gene versus they're doing really well because within a gene they're doing uh they they can identify the damaging um uh mutations and so what we did is for each one of the methods we converted the method into a gene score just by taking the median score in the gene so now we're going to say uh for each method it's got its high-scoring genes and its low scoring genes and just on the basis of those Gene scores how well will they Do
these methods do um on these exact same benchmarks so here the red is the same data from the previous slide but now we've broken it down so that the blue shows you the performance you would get with only the genan score from the same method um and to me this is actually pretty surprising to see that uh the gene score in is not a huge component of accuracy um for for denovo variants um another thing to note is that the relative performance of the Methods is really different it's not the case that the methods that
are doing really well at pathogenicity prediction overall are the same as the methods that are doing really well um at uh at Gene scoring uh in fact you can see that MPC uh is the method that does the best at Gene scoring across the board but of course there's a need to um go further and look at methods that were designed specifically for Gene scoring and start to compare them and see um uh both Benchmarks the performance and then also I think that this shows that there's a lot of room for potential Improvement um uh
in this space but this figure still doesn't get at this really important question of which are the methods that are doing the best within Gene there's we have for example there are a lot of different applications so you might care about distinguishing disease variants from control variants for diagnosis but you also might be conducting a burden Test using exom sequencing data in which case what you care about is only within Gene performance um because you're never going to compare a variant from one gene to a variant from another Gene the tricky thing about benchmarking within
Gene performance is you need a lot of data um and so these data sets um are too small for us to start to build case control uh comparisons within each gene which is what we would like to do in order to get at this really important Task um and so we developed a new data set using a large population genetics database Nomad um but we used new data from Nomad and we were careful to exclude the data that was used in the training of these different um methods and so by excluding any site at which
variation had been observed um in the time period during which these methods were trained um we were able to come up with an independent data set that then because it's based on population Genetics data um uh had enough variance that we could do within Gene benchmarking um and so looking at this uh independent benchmarking using population genetics data um we could see that if you look again just at the overall performance the same methods that we're doing well in the heavy selection regime that are really picking out um variants for denovo disease for example uh
denovo variants for for um severe diseases are also doing well at This um task of differentiating between observed and unobserved variants in a population genetic database which is what we would expect to see but then if you take this same data and you now do within Gene um evaluations and you average overall genes then all of a sudden the results really shift around a lot um and in particular esm um which uh in our genomewide comparisons wasn't as competitive with the other methods as a within Gene method it does extremely Well um and I think
that we're going to hear more about esm uh uh shortly um so to conclude different variant scoring methods have different levels of accuracy at different parts on the selection Spectrum I think this is a really important thing for us to keep in mind as we're uh following this Super Active field um and there's also a difference between differentiating am among variant within a gene versus differentiating among variants across Many genes um we've released our data publicly and soon we'll be making our code public is Avail uh code publicly available as well and we're hoping that
that is going to make it really easy for future methods developers to Benchmark in a way that will make it easy for us to follow what's going on and how excited we should be about which Innovations um and then just to return to this um initial question where I started it's very clear now that AI is Extremely powerful for characterizing msense variants but this question of being able to differentiate between disease variants and control variants is a different question from the multi-dimensional characterization of what a variant does to protein structure or protein binding or protein
stability and so I think that a direction that it's really important to be focusing on now is um away from the the very well uh you know established prediction problem Of pathogenicity and towards the much more difficult and and complex problem of characterizing that function in all of its multi-dimensional complexity um so I'd like to thank the collaborators on this project um and I'm happy to take any questions well thank you for the wonderful talk um any questions we have maybe 3 minutes um just to keep us on time thank you wonderful talk so can
you comment uh is Possible if we can build a emble of methods you talk about various methods we can build a emble method and generative advisory network focusing on the misfeed data points so we can input R the performance of current available methods yeah I'm super excited about ensembling these methods I think the thing to think about is exactly what the task is that we're trying to do and what the data is with which we're going to train our Ensemble so for example one Thing I've been thinking a lot about is burden testing um and
there if you want to Ensemble method and specifically how are we going to Ensemble these methods for really powerful burden testing but what's tricky there is the optimal Ensemble is going to depend on the Gene and so I think that um trying to build a framework in which we can understand uh in a focused way what we're trying to get out of the methods and then figure out what kind of data we Have to supervise an ensemble be very careful about double dipping um then I think that there's huge potential thank you go ahead question
so uh so there is a very large gap um between a variant and pathogenicity you know encompassing like multiple length scales but um so do you think that collecting data at these different length scales and sort of perhaps like bootstrapping all the way up to you know an organism level Characterization which is like is this pathogenic or not is this something that you find useful yeah that's super interesting so I think that if all we care about is pathogenicity then an extremely useful type of information is evolutionary information and you see just a huge gap
between methods that are taking advantage of either cross species or within human um conservation or constraint versus methods that don't and so I think that in some ways that if if What we care about is pathogenicity prediction then that means we can skip a whole lot of function and and rely really heavily on Evolution um I do think that the better we can understand uh uh function or maybe what I'm saying is the only way to get there is not just to understand function evolution is is giving us a huge amount of information as well
I think that um the better we can understand function the better we'll be able to do pathogenic Characterization though there's a really high bar to be able to do something that's going to be informative on top of um a lot of these uh uh um evolutionary based methods but I think that pathogenicity prediction is not the only task out there and so to me the idea that you would be collecting a lot of data in multiple length scales and trying to characterize mutational effects at these different length scales is awesome in its own right like
Regardless of of whether or not that's going to help us with pathogenicity prediction we have this other task which is super foundational um and so I think that I'd be excited about that for those reasons thank you um so maybe one last question just for for the sake of time yeah this is a quick question on uh kind of the separation between coding and non-coding variants and if there's a lot of confounding between um you know conservation or other mechanisms that Perhaps are in your next uh next steps wait S I say that again there's
a difference between non-coding and coding variants right so as just discussed there's a um conation difference between uh but in in terms of your next steps and structural regulation that's where non- coding really CH yeah yeah so I think that non-coding variants are just much harder than coding variants and so I think that um to me there's kind of two different tracks of research and one Of them is let's take these coding variants because we already have such a big head start and let's run with them as fast as we can and then at the
same time let's think about these Foundation models for understanding non-coding variants um but where you know the AI is is not seen as much success in that area and even before we get to the AI methods we don't even know how to uh connect these non-coding variant to their target genes uh you know in a lot of cases uh Which is a problem that I have thought about a great deal we we're making a lot of progress but it's still something where it's not like having a msense variant where you can just say like oh
here's here's the first thing it does what's the next thing it does um so yeah I think we got to keep on working on that and that's just a much harder problem thanks all right thank you let's give one more round of applause okay so our next speaker is um Marash babadi um he's the director of the computational methods in data science platform at the broad Institute uh where he is also Institute scientist um he has done a lot of exciting work in the Single Cell space and he's recently very excited about single cell Foundation
models so today we will hear about his talk on salarian GPT and salarian CS so with that maras please take it away thank you very Much uh oh is this the slide advancer cool all right so excited to be here um uh so today I'm going to talk about uh two of the projects that we've done recently um but before that I want to start with a with a few slides of dogma and I'm going to be careful with not to spend all of my time talking about the Dogma because I've done that in the
past all right so you know since the Advent of the Single Cell technology you Know we've been building these cell atlases lots of lots of data hundreds of millions of cell cell State observations you know in across different conditions from different donors in health and disease so the question is that what do we want to do with all this data why are we collecting all this data how can we sort of put this data to good use um better use than what it was potentially intended for originally so I would like to sort of go
Back to another um project that generated a lot of data and that's a human genum project then you know there were like many many like you know sequence short reads all these reads were aggregated they were indexed and then the the final outcome of that was a faster file you know one long file with like three billion entries I'm definitely oversimplifying it but uh now we are now collecting lots of lots of cell phenotypes you know we have like Trillions of cells in living organism you're not even like nowhere near there but we're just collecting
lots of lots of data so the question is that what is the final product so I don't think that a final product of this is going to be a database or a file it can't be that the final product in my opinion initially is going to be a search engine so I can basically make all this data findable and then a foundation model and I have like very strong Reasons for saying this so the Human Genome Project it was a point estimation problem we wanted to basically sequence one genome find basically one point in a
huge space of all the possible sequences but whereas understanding human cell a eukariotic cell is all about basically understanding distributions or conditional distributions like what cell State can we have in this context given this genome or if you're interested in Dynamics you want to know basically how does a cell State change in response to a certain action given a certain context and given a certain genome so very different problems you know one of them is to basically just estimate one point the other one is basically just learn a dynamical system a cell is a dynamical
system it's not a static thing so and I think the search engine itself is also going to be a Band-Aid um you know much like conventional search engines are Band-Aids now and they're being replaced with AI based search engines so I think what we're left with is actually just the foundation model so my Dogma is that the final product of a successful human cell project has to be a foundation model and I'm saying it not in a boss wordy way in actually a very real actual voral way we really need to to do this is
the way to do this there's no other Way of doing it all right so now take it to the Practical so I've been involved uh in a project at the broad um led by Steve mccarrell and Evan mosco the human brain Pro variation project this single project is going to produce anywhere near like you know 200 million to 500 million U multimodal single single cell observations now if you're thinking about even like more ambitious projects like project radical that is going to Like you know look at like you know dozens of cells many many variants
across conditions do multiple readouts and probably is going to produce anything you know from 1 billion to 10 billion cells so I'm not an engineer but I realize that we actually need to have engineering chops so I basically put my engineering hat on about you know three years ago and uh I basically started leading a team that built this product Called the salarium platform so what salarium AI platform is it's a cloud native sort of software infrastructure for data operations machine learning operations that scales to billion scale data sets and Beyond um it allows sort
of you know storing and quering lots of data rapid model development testing these models and turning models into products into sort of services that the community could Use um the platform has like multiple things you know inside it there's a data storage layer there's a machine learning library that basically in which we Implement how we can train large scale models in distributed environments on lots of lots of data and there some microservices that allows to glue things together and ultimately everything culminates into building applications okay I'm not going to talk about the library itself it's
open Source it's documented I encourage you to go read about it uh but I'm just going to talk about applications so the first application is what we Call salarium Cass or the cell annotation service and this is a work that is led by federal grab muscles and Kevin at the data scien platform and it tries to address the first problem sort of the Band-Aid problem of building a search engine for all the cell data that be generating why is it useful well because We need to be able to contextualized cell experiments we need to find
like this specific cell state is it similar to something that I've seen before or is it something novel so that's sort of the problem that Cass solves um you might have used Google gole inverse image search that is not called Google Lens you're basically doing the same thing for CS so the idea is that when you have a new single cell data set you're sort of embedding each Of your cells into a vector space we have already done that for you know hundreds of millions of reference cells and then we're sort of doing a vector
search in that in that space um so what is basically enabled doing this project is sort of learning like highly informative embeddings you know over large large scale scale large amount of reference cells and now once every cell is sort of endowed with a number of similarity hits In your reference data set you can actually look at what metadata is associated to each of those cells sort of summarize it and return it back to the user that's sort of what we call annotation which is you know a generalized form of cell type annotation but cell
type annotation is certainly one type of annotation that we we can return so there's a lot of engineering Machinery under the hood to make this sort of a performance service that sort Of scales to you know hundreds of thousands of requests um I'm not going to talk about that uh one thing that I do want to say is that summarizing of what the community have said in the past about a data set or a or a cell is actually a very difficult task because people use different terminologies to refer to different things so it's very
important to use ontologies and sort of use the relationships between the labels to be Able to sort of summarize and build a consensus and that's also something that is baked in baked inside the sanation service so uh the way it works is that you know the interfaces that you bring a fresh data set all of that is happening under the hood and then what you get uh as a result is you know some if you're doing cell type annotation you get the cell type ontology uh with all the ontological terms that are relevant to This
data set with scores and relevance uh uh you know colorcoded and now you can select your favorite ontological terms and sort highlight the relevance of that term for every sell in your data set you can do this you can do this um we released a preview of this um back in July uh to a number of broad Labs um a public beta of this will be released next week and just yesterday uh 10x genomics they basically released a beta version of this uh in their sort of Cloud analysis software Su so this is work that
we've done in collaboration with with TX genomic um if you want to try it for yourself uh please just go to our website Arium a there's a weight list that you know shortly you receive an invitation link to to to use a service okay so let's switch gears talk about the second project which is a sarm GPT this is the one that I'm personally more excited About so the next Frontier um at the intersection of AI and C biology is to be able to do in silico s biology and what I mean by that is
to be able to let's say predict something that you haven't observed about a cell such as you know one missing modality from another modality or more interestingly to be able to answer questions such as how can you reprogram cells you in response to Certain interventions chemical perturbation the transcription Factor over expression a crisper intervention and the whole Grail of that is that how can we remodel tissues like how can we take sort of a fatty liver and you know predict computationally what kind of intervention would actually remodel this tissue into like a healthy phenotype very
daunting task um but this set of problems are very different from you know a search Problem this is you know context specific prediction task and requires us to sort of learn the language of cells and tissues um which is sort of you know the second thing that I you know inside of my Dogma that we should do about cells all right so we using Transformers to do this um and the reason is that Transformers are just superbly useful for this task for at least two properties among many other properties Uh one is that Transformers are
basically invariant to permutation of the input and the facts that we know about the cells these are not ordered like you know that this Gene is expressed that Gene is expressed you have this surface protein the chromatine is in this organization so these are like independent set of facts so if you want to sort of learn sort of a joint distribution of all these facts Transformers are really good for that The second reason that Transformers are useful is that in the same sense that the usage of language in natural language is very context specific and
context dependent in a sense you know the words sort of get their meaning from the nearby words the same thing can be said about the gene function and the function of Gene products and the way basically Gene products interact with each with each other to sort of modify the function of each gene it's very much Like how things happen at the natural language and Transformers are equipped with the right type of implicit bias you know the type of structure such as self attention to actually be able to to um it's you know to to learn
these things from data um so what we' have done is that we've basically taken lots of single cell transcriptomics data and we have built a vocabulary for this data and this vocabulary uh includes context of This experiment that this cell is coming from uh and also the observe cell state in this case just transcriptomic cell State um so we we still haven't really done anything the space of really connecting this all the way back to genomics and in my mind that's you know the the next Frontier um and then what happens is after you build
this vocabulary is that you know all of these tokens will be embedded very much like how in naal Language you know text pieces are being embedded in and then everything goes into a Transformer stack and then we make a bunch of predictions such as you know predicting some of those context dependent labels and also like predicting um the the count distributions so there's a lot of details that goes under like how you actually train these models efficiently and you know what type of regularization you use to to basically learn and uh I'm We're going to
have a pre-print on this soon but let me just show you some results so the very first result is basically just to check how much Immunology your model knows about like does it have like basic grasp of hematopoesis so what we've done is that we've just taken two cells let's say like a naive cd8 positive T cell and then affector memory cd8 positive T cell and then we looked at what are the genes that are like commonly expressed in the Two cell types and what are the genes that are only exclusively expressed or like more
highly expressed in the affector cell state so we prompted Solarium GPT basically with a series of prompts and ask a series of queries so the prompt is that basically in we start with just the common genes add one at a time and then we start adding those like affector genes and then we asking you know for the query we asking again some genes That are in the fector state and some genes that are in the in the naive State and here's the result so like nkg7 and granzyme a these are two genes that you expect
them to be Express expressed in the affector state and we can see that the moment we start prompting the model with cattin 7 then the expression level of nkg7 goes up and also the same thing happens for Grand a and it continues as we just prompt the model with more and more of the affector genes likewise if You look at for example tcf7 and lf1 that you know are genes that are like just generally lower Express in the naive State we see that as we basically tell the model that you know this is an actually
a vector- like State the expression values of these things go down the other very cool thing is that if you just look at these distributions and these are distributions of copy numbers of mRNA they very much look like mixtures of negative binomial Distributions but we never taught the model that we train the model in a fully non-parametric way but the model has actually learned from this distribution of counts in the single cell data sets that negative binomial distribution is actually a good distribution for describing the the outcome of the experiment this is super cool uh
all right so the next thing we did is that we basically asked that okay so now that we have this model how can We actually extract some biology out of that so what we did is that we performed a series of experiments we just take one cell and we take one Gene and we just reduce the expression level of that Gene and leave every other Gene the same and we repeat this for every other Gene and we give these as prompts to the model and then basically query what the model thinks the expression levels of
every other every Gene should be in the cell so what we would expect to happen is That if you know there is some Cor ation or covariation sort of is going on between a group of genes since every other one of those genes are left intact they will sort of pull up the expression level of the gene that you sort of knock down but the gene that you knock down will pull down the expression levels of the genes are sort of covariant with it right and then so what we can do is that we can
compare the output with the Baseline and sort of calculate some lock Fold change with respect to that Baseline and we can think of these outputs as sort of a a covariation signature of every Gene and now we can build a gene Network out of that based on the cosine similarity of this Co variation so let's just see some results so we took one Mighty cardom myosite that we have a lot of it in our training data and we basically did that to it and this is sort of what we get out so in this plot
every point is a Gene and the points that are Sim close to each other these are basically genes that have similar L Co variation structure okay so we clustered it but now the question is what do we do next so Churchill has this very famous saying that Gene set enrichment analysis is the worst form of single cell analysis but it's the best but it's you know except for all the other forms that we've tried um so that's what we did we basically took every one of these clusters and With the gene s to enrichment analysis
to see whether these clusters corresponds to one of the known genes and lo and behold it does so that tiny dot over there if you zoom in these are all of your mitochondrial gen if you zoom in on the down below these are all the ribosomal subunit genes if you look like right there these are sort of uprs if you look down below these this is the electron transport genes and Then if you look in the middle you see the cell cell Jun function organization genes and just right somewhere in the middle you see all
the genes that are involved in muscle contraction and ccum your organization that you expect to see in cardom myoides and also myofiber assembly so we basically see core functions we also see cell type specific functions just popping out of this model that is trained on lots of lots of observational Data so we tried something else we also basically tried to give this model an H3 immortalized RP V cell line and this is something that that is not in our turning data set this is just a new cell type so so far as the model is
concerned we repeat the same experiment and this is the map that we find again if you look at you do the same sort of clustering and enrichment analysis we find again up there these are all the r ribosomal genes here we see cell cyle Genes all of them every like this whole like left Island these are cell cycle genes and this is very important because these immortalized cell lines is cycling cell lines and so that's exactly what you expect to see among other things the other thing that is very important is that salarium GPT had
never seen cycling RP one cells we only trained solarum gbt on primary cells and primary RP cells retinol EP pigment epithelial cells these are all postotic they don't cycle But if you give as input put to the model a cell that looks like something that has you know was in the training data set but it was cycling and Salam GPT understood it in a zero shot setting and this is in my opinion an instance of basically in context learning you know just basically observing something new and you know sort of being able to synthesize facts
about it from from the rest of the data um so the other thing that you know We did is that we um sort of try to compare the output from C mgbt with actual crisper perturbations but you have to be careful about that because what we see in Sal GPT these are basically covariation structures that in observational transcriptional States and so you can imagine that there's sort of homeostasis manifold for all the cell States and then you have oxid stress stress Contex you have like hypoxic contacts you have Different D genetics cyto stimulation transcriptional fluctuations
and because of all these sources of variations the gene expression sort of you know lives on some type of a manifold and that's sort of what sgbt is picking up on but an actual peration experiment is a bit of Beast because you're basically acting on a cell and then the cell can potentially even go off of the homeostasis manifold so in a sense it's sort of comparing apples to oranges so We don't expect right now s mgpt to be able to predict crisper perturbation responses but nevertheless we tried that so under right you see basically
the same gene network from an actual crisp perturbation and on the left the same gen network from that I show just you showed you before we can sort of try to color them based on the spent correlation of the responses so a blue Gene is a gene that the covariation pattern that we seen C GPT is compatible With the crisper response and a red Gene is a gene that is completely incompatible and you know this is we're just starting to look at this more closely but what we see is that you know cell cycle DNA
dependent DNA replication anything that is to do sort of it U sort of cell cycle these are sort of the same between the two setups and we believe it's because a crisper knockdown of a cell cycle Gene arrests cell cycle and brings down every other cell cycle Gene Together with it and that's sort of why we see compatibility but if you look at some core cell functions such as protein synthesis mitochondrial translation elongation nuclear export the crystal response is very different from from a covariation structure of you see and because if you just knock down
one of the ribosomal subunits the cell dies eventually right okay so quick conclusion Outlook um so Salarium cast makes about 100 million public available single cell data inverse searchable and you know totally encourage you to just go on a website and sign up for the service and try it out and give us feedback salarian GPT um I think it has achieved a remarkable grasp of cell type specific cellular processes and we will open release all the code and all the model weights U as soon as you know in a good shape to do That um
more generally I think AI really presents an unprecedented opportunity for cbard research like we have all the evidence for that now the other piece of evidence that I guess we have is that top down approaches are better than bottom up approaches this is something that we've seen in natural language we didn't learn to synthesize text by starting from grammar and syntax we just fed a lot of text to the models we didn't sort of learn to design Pro Proteins by doing like quantum physics and simulations even though we should do that probably uh we basically
just did you know lots of lots of you know cryoem and train models and just you know that that scale I think there's there's a good philosophical and you know and physics based reason for for this because every system has its own level of description at different scales so cells has its own sort of emerging level of description that you can basically Learn cell function but just looking at cells at that scale you don't need to go and really understand mechanistically how every single protein interacts to be able to say something meaningful about cell states
of course you want to be able to modify cell State that's a different story so that's need that's why you need to ultimately connect all of this back to biochemistry but I think we can make a lot of progress in understanding cells and understanding How cells respond to to interventions just by by looking at cells at sort of you know at at the at a high large scale lens um the outlet look is that we need to standard develop standardized quantitative benchmarks for all the cell language models because that's how you can show can make
progress then fine-tuning sgpt on various prediction tasks such as you know chemical and Crystal pations viability screens so on and so forth and Then perhaps you know calibrating the confidence of these types of cell language model predictions towards sort of making progress in the labing the loop Type U scenarios so that we can use these models sort of guide the guide generation of new data sets and ultimately I think the whole Grail problem is how we can marry biochemistry and structural biology models with s language models that's one of the hardest Problems so with that
let me just get to my acknowledgements so great great thanks to salarium GPT development team uh Steven Fleming and Yas ORF uh specifically like fed grab for salarium cast folks on the data Sciences platform broad legal Eric Lander so so uh I've been meeting with Eric Lander we have been meeting with Eric Lander for the past two years and uh We've benefited so much from from the Guidance that we see from Eric Lander I also lost half of my hair I think it's I think it's worth it I still have more hair Eric so just
um Steve McCarol from uh the center for human brain very ation for supporting a lot of the works that you're doing on a grea for super inal discussions 10x genomics for supporting uh cell anation service CCI cell by Gene project for making a lot of standardized Data available to the community and a lot of the interesting things that are happening right now is really just owing through them for like really standardizing all the data so we can actually do machine learning on that data because otherwise we just couldn't we had to spend all of our
time curating the data and lastly folks from cerebras who gave us access to their wafer scale um GPU well wafer scale devices which are some of the most marvelous um pieces Of chips that I've that I've seen in my life so we've been sort of training all of our models on on C grass and finally like folks from the Argan National Lab for allowing us to sort of use our Computing facility thank [Music] you all right thank you for the wonderful talk uh actually we have been talking a lot about this face as well and
I should worry about my hair uh we are running a bit short on Schedule um but maybe reach one or two questions real quick so we can give Alex more time hi thank you very much for a very interesting talk um just a couple of quick questions so do you Recon this um do you reckon we can this um salarium GPT can potentially be used for example for our identifying Co varing genes to develop probes for targeted a special transcriptomics and also is there do you see an opportunity To uh study these core variation to
take this core core variation embeddings and uh apply them in context of case control data sets to see what groups of genes are coing between like Disease and Control yeah uh both of them are great questions the answer to both of them is yes so definitely you know you can identify you can basically start with a gene and you can use the co varation pattern to sort of extend it to a larger Gene set and perhaps you know some of Them are better targets depending on the asset that you're designing so definitely that and yes
so the the responses you get from salarium GPT these are like cell State specific so a very intriguing question is to basically just see you know what would be the difference between the gene Network that you see you get from you know the health healthy State know wild type state to sort of a disease phenotype so we we are in the process of doing these types of Ttings but I don't have anything concrete to share at this point um maybe let's give a chance to the folks in the back they are harder to come up
stage uh we can definitely accommodate your question offline yeah let see one hand in the back or did that me something sorry okay go ahead quick question can you use GPT to create biologically meaningful EMB badings for single celles or C lines Yeah great question so the the problem of like embedding is is is a is a very loaded thing so because why do you want to embed something because if you want to embed something to be able to find similar stuff in that sping in embedding space but what is similar stuff so do the
two cells you know every cell has like you know billions of like well not billions but like you know thousands of different functions right so you might be actually interested in an embedding That emphasizes on like an inflammatory response pathway right so there is no single embedding that you can make so I basically want to want to say that maybe that's not the best question to ask even though we can get like you know million different types of embeddings or whether this embedding is better than somebody else's embedding it really depends on you know what
task you're subjecting these embeddings to so perhaps a good maybe a great question which um I'm Inspired just by your question is to basically ask like can we use like you know stuff like salum GPT to build embeddings that sort of emphasize on like different aspects of Cell Biology and perhaps we can yes I just thought the model structure was a bit similar to The Universal Bing model from Ure Le or the recent SC Foundation model that was in nature so I thought maybe it possible to to to to use it for T presentations yeah
yeah um so yeah so There are a number of you know similar uh efforts in the in the community uh you know scgp G former SC foundation and again from each of these you can get embeddings but again if you don't guide these embeddings into the directions that you actually care about for the downstream biology that you're doing the quality of the embedding is just a toss up it's basically just based on the you know biases of the model that you have so you can get embeddings and use them For different tasks but I think
I would start from the task and then go ask how can I make an embedding that is useful for that task thank you okay um um thank let's give marach one more round of [Applause] [Music] applause okay so it is now my real pleasure to introduce our second keynote speaker Alex Reeves who is um very soon going to join us here at the broad as a core faculty and also the department uh Of electrical engineering and computer science at MIT so we're we can't wait to actually have you here whole time so it be really
exciting um so Alex is an expert in developing AI models um very broadly for scientific discovery for scientific understanding for scientific design in biology um and he is currently um founder and chief scientist at evolutionary scale um and I hope many of us have seen in June um the new model of esm3 being released and I'm sure you Will be telling us much more about it so welcome Alex and the stage is all yours all right thank you very [Applause] much there we go okay well I'm going to be talking about um our work over
uh a number of years now so I guess about five years developing language models for biology um and so through large scale Gene sequencing efforts um we've collected the sequences of you know billions of Proteins across the tree of life and you know if you think about these sequences they're written in a language that is incomprehensible to uh the human intellect and so kind of the question of of esm research program is you know can we learn um biology can we learn to understand um these sequences through language modeling and we have the techn to
read um and print genes and so you know we have biology is fundamentally programmable at this very basic level But in order to program it and to go beyond sort of taking the parts that biology has already created and repurposing them we would need to understand this language we would need to understand how to read and write it um and and there's you know just a tremendous depth of of complexity there you know it's a language that's that's prior to all other languages and that um ultimately you know um distills down into basic uh biochemical
and physical Principles so the methods that we're going to be using are you know these incredibly simple methods um from a certain standpoint um and so you know one one might wonder how could this possibly work um and and at this point probably we're all familiar with what language modeling is but but just to remind all of you how simple a thing this is um one is given a sequence and one trains a model to predict tokens in that Sequence and and kind of the classic approach is autoaggressive so um to be or not to
be that is the the blank and the model is asked to fill in um the the next token given all the proceeding tokens another variant of this um that that is is also important is mass language modeling where a number of different uh positions throughout the sequence are masked out so to to blank or not to blank that is the blank the model is asked uh to fill in those Tokens um and and so the the training objective for these models is is very very simple and you know what we've seen across artificial intelligence um
and and I will hope to convince you that this holds true in biology as well is that as we predict um tokens across very large data sets which contain um patterns that reflect sort of a deep structure um the models can discover uh this structure and and so that is the core idea the core Hypothesis behind esm and if I have time at at the end you know I'll talk a little bit more about why we might um think this could work but um you know I want to begin by by convincing you that it
does work and so if you think about Evolution and sort of the ba vast you know scope of of of time and the immense set of experiments that are conducted by evolution in parallel and our ability to read those out through Gene sequencing measurements we have this this view of Biology but only through the process of natural selection which tokens Evolution has chosen to place in sequences um and this idea dates back a very long time that if we were to look um at the patterns within protein sequences and particularly within individual protein families um
you know we would be able to see um statistical correlations which would reflect the underlying biology and um this this is shown here it's an idea that's fundamental uh to Alpha fold to Methods of protein structure prediction that predated Alpha fold um and for example if two positions in a protein sequence um which are uh far apart um from each other in the sequence but close in three-dimensional space so that they interact in some type of tertiary interface um spatially uh you can you can imagine that evolution is not free to choose their identity independently
and so um because of this Fact um if evolution switches the identity of one amino acid it would have to um make a change in the position that's in contact and so we can see these patterns and essentially you know as soon as we were able to begin um sequencing genes at a at a scale enough to look at multiple members of a protein family these patterns became visible and so you know at a very basic level one can um imagine that it is this sort of thing that um that a model might uh Begin
to learn about and so um you know we began looking at this uh and and we we put out a you know the first first pre-print describing esm in the spring of of 2019 and you know this is the core idea which is basically we're going to train you know large Transformer models across um databases of of of millions of hundreds of millions of protein sequences and um we'll ask these models to predict tokens across Evolution and then really look at what happens as the Models learn to solve these tasks and what um you know
what representations what properties develop and emerge within the representations of these models um and so uh one of the one of the first things that you see that's I think very interesting is the emergence of um protein structure in an interpretable form within the attention maps of these models and so what is shown on this slide on the um on the left hand side uh the the Transformer Proceeds through um a mechanism that's called self attention where each position in the sequence is allowed to look at every other position and sort of a weight is
assigned to how much information will be transferred from um one position to that Central attending position so you can imagine this as kind of synthesizing a graph along which information will be passed and and and so as as as as um as the input um you know is processed through the model um Block by Block in each of these blocks um self attention will be performed and and for each input a different graph will be synthesized and so you know the question is what is the structure of this graph and so you can look at
um the attention maps and basically you will see that there are actually a set of um attention heads which across you know many different proteins which which differ greatly in their biology and their structure um there particular Attention heads which will um correspond to contacts uh and and long range contacts in the three-dimensional structure of the protein and and this is of course has just been learned from the sequences so these Mongol haven't been trained on structure they've just been trained to predict amino acids and you know one reason for this might be um the
the uh the the logic um that that I described in the previous slide um but but on the right hand side you can Basically see um a map of the attention heads in this model so you know basically the bottom is is the the input layer of the model the top is the output layer each of the heads are are are across the the the x-axis there and so basically you know there the the set of these attention heads that correspond to long range contacts and and interesting they're in the the later layers of the
network so they're kind of synthesized later On um you can actually take this now and fit a really simple logistic regression basically to predict the contacts and you get something that is is is uh you know essentially a state-of-the-art unsupervised uh contact predictor from this so um another thing that's that's really interesting is um you can look at the probabilities that are output by the model so it um you give an input sequence and the model will uh give a probability for each token within that Sequence and so um you can look at the uh
uncertainty of the or or the entropy of the uh um prediction for for each token um and you can look at which uh tokens are predicted to be uh in each position and we also see that there is a correspondence with biological function here and again you might consider this unsurprising in the sense that um you know the the uh the the to tokens that are peer have been selected by Evolution um which tokens are are feasible for Evolution to place are tokens which are consistent with the the function of the protein um but what
we can see here and this is a map of the model's entropy and so if you just take the and I believe these are the 10 um lowest entropy uh positions in the model's prediction shown in blue here on a DNA methylase these are clustering in the active site of of the enzyme and in the residues that uh interact with um with DNA uh so you can look at this more Systematically and and that is shown here across a data set of deep mutational scanning experiments and so the x-axis different uh deep mutational scanning experiments
you know some of these have uh have hundreds to thousands to to I think hundreds of thousands of um mutations in them depending on the data set and we're just looking at um a log odds ratio so sort of the the the the log odds ratio of the a mutated uh amino acid um as estimated by the model Versus the the wild type as estimated by the model and um you know this this is then a score for um the effect of of mutations and and you can just look at a Spearman correlation with the
actual uh functional scores here so this this is again completely zero shot you know we're not supervising the model with any information about function we're just you know constructing a score based on the probability that it assigns to each amino acid in the Sequence um the other thing that we saw this is from uh ladol in um uh this was pre-printed in the summer of of 2022 um we trained a series so the first esm model was a 700 million parameter model mod and so at the time that was a very large language model um
it came out uh you know very recently after uh The Bert model and um gpt2 which were kind of at a similar level of scale and so we're very interested in sort of what happens as you scale these these models Up and and one of the things that we saw even in the first esm model is if you train models at different scales um you can actually track sort of a correspondence between the perplexity of the model which is measuring its accuracy um in its token prediction task with um the emergence of biological information such
as structural information and you will see over the course of training as the perplexity goes down that the structural Information increases and also that um as you Scale Models up they can attain lower perplexities and uh the structural information that they learn will increase and so we saw that up to 750 million parameters uh here we um we train models uh from 8 million parameters to 15 billion parameters an entire family of models and um we looked for the signature of atomic level structure in these models and so what I'm showing here is is um
a very simple Thing it's basically um you take the the model after it's been trained the representations are frozen and now we're fitting um an equivariant projection head onto this model and that equivariant projection head is supervised with um protein structure data um and it's outputting uh coordinates for each of the atoms um in in the uh in the protein there are no folding blocks there's no multiple sequence alignment um you know this is a Very just simple projection and so what we find is that um there's enough information in these representations that you can
um predict you know project out in many cases um a high resolution structure for the protein and um this information uh improves with with scale and you know this is just showing one example of this on a test set protein um one of the things that that we see I I don't have the plot for this but you know it's in The paper is that at each jump between model scales you know a diff proteins will be learned at different scales and so harder proteins will be learned later and so you can see there's kind
of um a progression uh where this protein is is learned and actually it's learned relatively early here between you know primarily between the 35 million and 150 million parameter model okay um we've open sourced these models one of the I I just want to highlight to I think really Nice uh papers that uh we didn't have anything to do with but um that that have explored ways that you can use these models um this is a study by brandol that looked at predicting the clinical effect of mutations and so you know taking those um log
odd scores and and basically using them to score variance across the human genome and they were able um to find uh that that uh es m1b and you know without using again any kind of multiple sequence Alignment or anything just kind of using using those raw scores um was was a state-of-the-art predictor of mutational effect um this is a paper by Brian he and and his team on optimizing antibodies with esm and again this is really just using the uh the token probabilities of the model and um it's a really exciting paper they were able
to improve the an uh Affinity of seven uh different antibodies um they measured fewer than 20 new variants of each antibody across two rounds of Evolution so this is an extremely efficient process and you know they're really just scoring these these uh potential mutations um uh using the the probabilities um that esm outputs these representations can enable um very powerful predictive models uh so um as we built esm2 and we saw that um there was an atomic level picture of protein structure in these models you know the the natural question is can you Use this
to do um a single sequence structure prediction can you really kind of go um from from a raw sequence without uh multi multiple sequence alignment information you know directly to a 3D structure and and basically using a very simple head using um a series of uh of blocks that have a similar kind of pairwise inductive bias to Alpha fold um that use triangular attention and some of those uh key mechanisms you can put that on top of Frozen esm2 embeddings and get a state-of-the-art uh predictor of uh Atomic level structure and that's shown here um
on the Cameo test set accuracy uh you know approaches that of alpha fold but um the inference is is much faster here because we're not um using multiple sequence alignments you know we're just uh you know we're just basically putting the sequence in and using esm to uh produce embeddings and then project those embeddings into Coordinates um there's also uh generalization that that you can see where where for instance the model can learn um to predict interactions between uh different chains even though it's not been trained uh explicitly to do that so you know one
of the big questions I think that people have had the scientific Community generally has had I think that just naturally emerges from thinking about this predictive task is you know can these models Generalize and it's a question that's been asked about language models um in in in diverse domains not just in biology but more more generally in vision and language it's very hard to ask this question rigorously um in uh in natural language because there is um there's a sense in which the evaluation has an inherent subjectivity um but in biology of course we can
um we can query nature we can we can look at this question empirically And so um you know this is a picture of of protein space it's it's one which um uh uh David Baker and pasu hang have um have uh have proposed a picture like this to think about denovo protein design and so you can imagine the space of natural proteins which Evolution has explored um occupying uh you know a very tiny fraction of the space of all possible uh proteins and if we ran the experiment of evolution again you know it may may
end up on almost certainly Would end up on a different set of of proteins um and so you know can these models really go beyond the set of natural proteins that uh Nature has has created for us already and of course if they couldn't they would be I think much less interesting um so you can look at this question rigorously uh by asking you know can these models uh create proteins that that are that are distinct from that really go beyond this space that don't fall in existing Natural protein families and so we look at
this in two different ways the first way is we uh take a set of uh protein structures which have been created by denovo protein designers to differ from natural proteins in ways that they consider meaningful so this could be different topologies or um you know changes changes in in the shape that that are meaningful and that um they consider to to to really be distinct from um the structures that are Used to realize those those functions Naturally by Nature so so that's that's one test set that you can look at the second um way to
really look at this is to just generate generally from the model and look at the sequence similarity to um existing natural proteins and really ask you know can the model find things uh that that don't map to existing protein families in sequence space but which um but which do do fold so we look at this question in both of Those ways so the first uh the first slide here is showing um the evaluation uh when we take a set of uh denovo uh structures that have been designed by protein uh designers uh there's a test
set here of eight different structures um we uh we expressed these proteins we looked at two things we looked at their solubility and then we looked at um uh size exclusion chromatography and um um uh the the the appearance of a um of of of of a you Know a Peak at the expected molecular radius um for for the design um that's that's uh so so you can look at both monodispersity or uh or not you know non- monodisperse proteins which would uh you know which which might be occupying more than one um State they
might be diers or triers or so on um we're not explicitly designing for monomer so we're considering it a success if we see a Peak at the right molecular radius and so we we see a high Success rate and and one thing to mention is that uh protein designers have seen this uh correlate with um you know the the uh the existence of well well folded uh protein so um you know here we see a very high success rate just for Designing for these backbones what we're doing is basically inverting the L language model so
we're projecting out this low resolution picture of the 3D structure that I showed you before and then and then we're optimizing the Sequence so that that um contact map matches the contact map of the denovo protein backbone um and so we we see an 80% success rate here um for these uh designed uh backbones so now this is looking at kind of the second way of of doing this which is um which is generating uh sequences and and so here what we're we're we're we're optimizing we're performing this kind of joint optimization where we're looking
for um uh uh sharpness in a sense in both the The structure and the sequence probabilities and so we're really looking for sort of a joint optimality between the sequence and structure and so as we descend an energy well so we're basically um alternating optimizing the structure and optimizing the sequence the structure optimization again is just in this projection space of the contact map you can actually see that in the animation on the bottom that just ran and so we descend into an energy well at The bottom of that energy well is um a protein
design and so um this is a map the proteins are colored by their sequence similarity to um the nearest natural protein and so you can see that um the distribution of these design both um overlaps with and is distinct from natural proteins and that there are um a diversity of different structures that are produced by the model and again if you put this to the experimental test um we saw 55% uh overall success rate by size exclusion chromatography and so finally you can ask the question okay so out of that set um if you look
at the set that is really distinct from natural proteins and so that's shown in the lower left corner here um these are these are proteins which have a uh sequence uh identity less than 20% to any natural protein for uh a number of them that are kind of uh at at the zero point on the x-axis um you know there's no Significant similarity that can be detected and then if you take each of them and you fold them so you get a predicted structure and you search that um against um the uh the natural um
protein uh database now now we're looking at the structural similarity of of the nearest match on the y- axis and so these These are proteins which are um distinct in in sequence and structure and here we also see you know a 63% success rate so you Know these in in a certain sense these Generations are no different from the generations which are much closer in sequence to uh natural proteins um I'm excited to present this this is a uh crystallography result so this is for one of the fixed backbone designs um and here we've we've
uh we've been able to solve uh the structure for um uh f00003 which has um a sequence identity of 22% uh to the nearest natural protein actually I think this is to the template design so because this is one of the fixed backbone designs so this is a um a protein which denovo protein designers have created with a new topology and so the model has found sort of a distinct solution from that that which protein designers found but which folds to this uh structure so I think it's interesting to to think about the implications of
this And you know maybe maybe talk more about that because there's there's some form of very interesting generalization that's happening here I think I think you can't conclude from this you know if if if you're if if if if if if the starting thought was that these models would memorize the existing protein families I think that this contradicts that um something more interesting must be happening And you know what that could be is sort of an evolutionary grammar which links sequence to structure um there may be there may be more than that but I think
um at a minimum you would have to conclude that there is something Beyond sort of a memorization of the existing protein families okay so I'm going to switch now and in the remaining time talk about esm3 so esm3 is um our newest model uh it's say it's a model that's been Trained now on on another uh order of magnitude in scale um and so we train models uh from 1 billion parameters to um close to 100 billion parameters on uh billions of protein sequences uh esm3 is a uh a mass language model but it differs
from esm1 and esm2 in that it's a multimodal mass language model so it's a model which reasons over sequence structure and function and it's been trained with a generative Mass language modeling objective and so instead of Being trained just to predict tokens that are masked out at 15% of the sequence um it's been trained to predict tokens across all possible Mass grates so because of this you can generate a sequence using the chain rule of probability so the model has been pred uh trained to predict the next token given an empty context given one token
given two tokens given three and so on all the way to a sequence that only has One token missing and so essentially you can generate any token in any order and um generate a sequence from that now it's also interesting about this is it makes it prompt because you can start the context with anything and so if you're interested in conditioning the model on a certain structure or certain functional Motif um you can you can put that into the prompt and you can ask the model to find completions they make sense uh and so um
this has has Connections to um diffusion discrete diffusion models it's it's not um a discrete diffusion model but the uh the the the the the chain rule factorization um is uh is something that ties those two approaches together um so kind of the key Innovation that enabled this is the tokenization of protein structure and so you know basically being able to convert uh three-dimensional structure into discrete tokens that a language model can um reason over and so to do This um we cre created a VQ vae Auto encoder um that basically Maps local um local
spatial neighborhoods in a protein into discrete tokens um that uh compress their geometry the decoder is then trained given a sequence of tokens to uh project that back into a full three-dimensional structure um this is enabled by a geometric attention mechanism and you know this mechanism is essentially A variation on self attention that is Aware of the spatial relationships and so attached to each um position in the sequence is a reference frame and so um coordinates can be input locally to their reference frame and in the attention mechanism they're mapped into a global frame where
they spatially interact um and so they can interact through dot products um so through similarity in their Vector directions and they can interact um through distances so that Close or far positions can communicate with each other this can be um very efficiently realized Through The computational Primitives of attention so this is what the vocabulary looks like and um you know it's it's it's really kind of capturing these uh structural motifs that are local in space and so you can see that it captures um it captures both both both both local and uh and terer motifs
um the uh decoder uh delivers um Very strong reconstruction and so uh for for most of the test uh set we see um for for the training of the decod we see rmsds uh below one angstrom so you know you can you can really compress um the structure using these tokens uh so as I said we scaled these models um up to 100 billion parameters um the largest model was trained on 1.8 uh trillion tokens overall um over 1 time 10 24 flops of comput so a TR ous amount of compute uh that went into
Supervising this generative model and the data sets um include uh metagenomic sequence data um the protein Data Bank so so real uh protein empirically determined protein structures and then a large amount of synthetic data as well um using predicted structures and uh Inver folded sequences and you know you can think about this synthetic data I think there's something very interesting about it because in one of the directions you Go from synthetic to to real and so the model is supervised to predict a real sequence using um synthetic prompts and so I I think there's there's
some very interesting parallels to um ideas of natural language processing about around back translation where um you know you can use sort of a low resource um uh language um and you can make a lot of predictions and you can you can improve um the ability to do translations and and so you know using These types of principles I think I think it's possible to this is a very rudimentary kind of form of programming to kind of generate these prompts but you can imagine generating much more complex prompts synthetically so I showed you the the
map of proteins with esm2 this is the map with esm3 unconditional Generations you can see there's a much higher level of complexity the distribution both overlaps and is distinct from uh natural Proteins in sequence space the right hand side shows histograms of similarity in sequence and similarity in structure and um what we can see is when we generate unconditionally um the generations are similar to the Natural distribution and structure um they they are distinct in sequence when we um but but what's interesting is when we begin prompting the model we can steer it away from
the natural distribution and so here are shown two different types of Prompting one is for outof distribution folds so these were folds that were not in the training set the other is using these very idealized symmetric proteins and no natural protein looks like them they've been created by protein designers and so you can you can push this model and it will generalize out of the natural space I think the the thing that's very exciting about this model is the the the prompt ability And the ability to compose prompts and so here we looked at you
know just very systematically you know can the model solve compositions of prompts that combine a highlevel instruction about the the the fold of the protein with a very lowlevel description of a specific structural Motif where we want to achieve an atomic accuracy in materializing that Motif somewhere in the protein so it's a scaffolding problem where we're saying you know Scaffold this Motif hold it in exactly this uh a particular um Atomic level configuration and and place that you know in a way that's consistent with a higher level instruction about the topology of this protein um
and so this this is an example of this you know to make it concrete uh you know could prompt with an alpha beta hydras and then um ask the model to uh to to to design um um specific set of atomic coordinates so you know the model can do This uh systematically um and that is shown here a variety of of uh of of prompts um and combinations of prompts and um in in many cases here um the the so so these are all good designs where the constrain rmsd the atomic um level rmsd between
the prompt and the design is is a good match um and for we searched these motifs um to see if we could find a significant similarity to other proteins that Contain the same Motif and and for many of them so the serotonin um calcium proteas inhibitor and MCL one inhibitor binding sites all of those we couldn't find a natural protein that kind of had that same topology and that Mot teeth um the other thing that you can do with these models is you can align them and so you know I began by talking about the
distribution of evolution but when we're thinking about designing proteins um you know Evolution hasn't uh Um evolution is is not thinking about um proteins which are not natural and new functions and so um we can't I think expect the token probability to coincide with arbitrary functions that we might like to design um so in um in in NLP more broadly there's the idea of alignment which is really how do you um take capabilities that have been learned by the model that are kind of part of the distribution of the model in some way and elicit
those capabilities In the model's generation and so the way this works is you basically construct it's it's learning by example so you construct pairs of positive and negative examples and you'll essentially supervise the model to put more likelihood on the positive examples while remaining close to the base model and so using this you can fine tune with a very small number of data points so here we look at alignment to um prompting Fidelity basically so now We're looking at very challenging tertiary coordination tasks where we have to have um parts of the sequence come together
and interact spatially in very specific ways and um we're defining a success is having a PTM so a a strong prediction of the backbone um above 08 and a constrain rmsd in that coordination task of less than 1.5 angstroms and the passet K is is basically measuring if you if you generate 128 um uh uh examples of this Tas or you you generate 128 diverse Generations um for for for each task what is the fraction that can be solved with 128 Generations so if one of them is is good then you'll get um you'll you
you'll get a solution at pass at at k um so what you can see here is that you know there are differences between the models and their Fidelity the base models and their Fidelity to The Prompt but when you apply this alignment there's a latent Capability that is is elicited and so um the aligned models can solve double the uh Atomic coordination task and so we we see this um this this potential that's not revealed in the evaluation of the base model that um can be elicited by basically a small number of examples that are
used to to teach the model um this also increases the diversity of the generations that's that's shown here the diversity before and after um and it shifts the overall distribution of the Generations the base model shown in in red in these density plots the Align model shown in blue you know upper upper left basically is um the the prompts that have both uh good crmsd and high PTM um so I I'm essentially out of time so I think I'm going to have to just very quickly mention um this final um part which is uh you
know we we really wanted to see can you know can we use this to to create new proteins um and so we we started with gfp I think Gfp is is interesting for for two reasons one um it's very easy to measure so it's it's fast it's easy to do and and two um it's it's a very beautiful mechanism that requires a very Exquisite fine-tuning of function um and as far as we know Nature has only invented florescence um one time you know all all fluorescent proteins kind of have have have this one um mechanism
and it's it's an autocatalyzed reaction so the proteins backbone atoms reconfigure Themselves into a chromophor so it's it's it's very amazing and so the prompt here is basically just those the core coordinating residues uh their atomic coordinates that um basically realize this mechanism um and then we ask uh esm3 to synthesize the rest of the protein um we use a Chain of Thought procedure so basically we iterate between optimizing uh the sequence and the structure to find um sort of jointly optimal Sequences and structures and so um we did this with the 7 billion parameter
model basically as soon as we had that model um you were able to generate a plate of 96 designs um and uh um you know these were bucketed so we we looked at things that were very close to you know AV gfp the the kind of the famous you know natural um gfp that was that was the first to be discovered um and and things that were were far away so you can see a lot of things that are Lighting up kind of in the lower right there that are pretty similar to to Natural um
avgfp but we what we're really intrigued by was this well B8 um where there's a very dim fluorescent signal here you had to be patient for it because it only appears on day seven um and it's 50x weaker than um natural gfp so you you know you would barely see it we weren't even sure is this here um but you know there there is some you you can see it as as shown on the right um in Comparison to a chromal form knockout and so we continued The Chain of Thought from that protein so the
key thing here is there's no kind of experimental data going in to fine-tune this model or to supervise a predictor or something here you know we're just kind of using the structural principle and we're asking the model to realize this structural Motif um and so as we continue that Chain of Thought and we made another plate of 96 designs um and and I guess I Should say about B8 why is B8 intriguing well it has um a uh uh a 57% sequence identity um to the closest member of any you know closest known natural fluorescent
protein so this is this is quite far away um and so we we contined that Chain of Thought and so on this next plate we got a few bright hits and the brightest is well C10 that's circled there and this is uh 58% Identity from the nurus fluorescent protein so so very distinct it's really the model has kind of jumped into to an island in sequence space that doesn't exist in nature um so there's 96 mutations relative to the closest natural protein or not the closest known fluorescent protein tag RFP um and the difference here
is kind of similar uh to what you would see between um um proteins belonging to uh different orders within the same Class uh so like the difference between Stone um Stony corals and cemes within anoa so if you kind of look at this and ask the question you know sort of a thought experiment like if if evolution were to generate this what would it take um and you look at you look at you know this Divergence has occurred over predictable time scales and evolution um you know this this would essentially Equate to something like 500
million years of evolution so I'll conclude there there I won't have any remarks on you know why this can be but but I would like to um I would like to acknowledge um you know the tremendous amount of work and collaboration that went into this um especially uh the work on generalizing Beyond natural proteins which is in collaboration with Sergey of chikov and a team at the um Institute for protein design and then esm3 was Created by this team at evolutionary scale thank you so much Alex for a really really wonderful talk showing us how
far you can push it by such clean models and really models where you're thinking about very carefully um and seeing what you can just get out from there and all the structures so we have time for questions and we'll have people moving around again and I don't really see but so maybe well do you want to start Yeah okay so thank you great talk um it's amazing to see the power of language model for predi structure prediction so From perspective of first principle it equivalent to solve a multi body short in equation so can we
conversely think that we can use language model of P prediction as to decompose this model as a simulation of quantum computation it's a it's a really interesting question and it's what I would have Talked about in my in my slides if I hadn't continued so I'll say a few things about this um I think it's it's a very different kind of simulator that we're creating here so you know the classic idea of a simulator is really to um to to to write down the world in basic uh basic principles and equations and follow their evolution
to understand you know what the what what are the possible future States um this is this is very different it's an Information theoretic model I guess it's a predictive model and so it's really looking at the correspondence between input and output um there's maybe some sense in which in the limit of infinite data these two things coincide I think the thing that I have found to be very um there there is there is an idea I think that has been behind the development of all of this which is basically if you consider consider the next
token it's it's generated by Evolution um and how does Evolution Select the next token the entire deep complexity of biology is behind that decision because it selects tokens which um do not lead to the collective loss of function of the system in which you know that protein uh participates so to solve that task well you would imagine that if the model can generalize if it can predict the next token with Fidelity it would have had to develop representations that are Capturing and modeling some of that deep biology so that has been the core hypothesis um
that that information would exist within the representations of the models and I think it's it's very important to think it's not in the token probability the token probability is the probability under Evolution but to solve that task it would have to develop a feature space which captures um captures the depth of biology now whether it whether it can go that far you know I I Don't know you know and and so I I don't I don't want to say anything about that that's an empirical question perhaps with enough data um it would it would be
possible but you know I I think there is a principle here where there's a distributional hypothesis of biology the data is generated by evolution there's a deep structure to it and the model to solve that task would learn something of the deep structure thank you can I follow up on one of these Comments because it was also brought up before in talks so in terms of like the synthetic data that you have used it still seems very small as compared to you know the full data that you were using for training so first of all
were you worried about using more and why maybe not using more um and then also the other question on you know that that's on the data side and on the you know scaling on the data side but then also on the scaling on the parameter Side which you've done a lot of what do you see in terms of laws that are emerging and where are you in terms of like do you need to build much larger models or is it do you see any tapering off as well yeah okay so on the question of data
um well we always want more data I think is the short answer to that question so it's never It's never enough um and we we want the data to be to be better so I I think you know the you Know the other reason for these these models of course is that there is so much sequence data and that's growing exponentially we can expect it to continue to grow we can expect it to Encompass so much more like cell States and we've heard about those models so there there is this tremendous growth of data we're
not seeing that same growth in protein structure data so there's kind of a limit there so I think predictive models are are really Exciting because they've kind of expanded you know our ability to understand structure to train these models I think there is some some issue which you you would expect to see some saturation because you know in a certain sense the model could learn to become a simulator of alpha fold with enough data like instead of becoming you know a simulator of the actual structure so I I think there's you know there's a lot
of deep Research to kind of figure out how do we use data augmentation um effectively here but but yeah of course we would want eventually to fold all the proteins in the training set and have sort of sequence and structure and functional data for for everything and then on saturation you know it's um it's we we don't see it and I I think you know and I think in principle like we we train this model in 2.78 billion protein sequences I think Um and so there are many more so it's you know I think that
you know there there are many many more I mean how you select them matters having good tokens matters um so so I I think you know I think it's an interesting question how how far we can go but I I guess I would say I think we can go further with the data that we know is is there thank you for this really interesting talk I was wondering um how does your model account for variations In sequence length like say when you're trying to predict the next token can that token does it have to be
one amino acid or can it be a deletion or can it be multiple amino acids so in principle you could do that but yeah the way this is set up is you would have to say how long the protein is um because it's not predicting an EOS token but you you could have a predict an EOS token if you wanted it to wanted it to do that but but esm3 hasn't been trained that way so You put in a certain context linkone when you're generating would you need would you need a lot more training data
to account for variations in sequence like than the model no no I don't I don't think at all I mean the model takes of course sequences of all different lengths because the Transformer can um you know would would would process them all it's it's not it's not a fixed context link so yeah thank You okay so thank you Alex for a really really wonderful talk I hope you'll be here for lunch to take on some more questions absolutely um and with this let's thank Alex one more [Music] time and we'll all be moving out for
lunch and meeting back here again at 1: 15 15 p.m.