transcriptomics three is of course dedicated to advanced methods of analysis that will allow us to find meaningful patterns in data especially complex patterns that are present in big datasets in this video we will turn our attention to supervised machine learning classification and feature selection in the transcriptomics 3 course we are learning about machine learning for transcriptomic data as we've already discussed machine learning can be separated into two main categories of methods the first of which is unsupervised machine learning which focuses on the detection of patterns and data the main idea here is to apply techniques
that are automated and rely on various assumptions about data in order to learn from it the second main category is supervised machine learning these methods rely on the availability of training data which has been labeled once the data set of labelled data has been accumulated it can be used to train a model that will be able to apply this knowledge to analyze new data for example by predicting its class large RNA seek data collections provide us with the opportunity to discover signals and gene expression that might not be apparent with smaller sample sizes such as
prognostic indicators or predictive factors this is especially the case for subsets of patients however discerning this signal in such large data sets frequently relies on the application of machine learning algorithms to identify relationships and high dimensional data or to cope with computational complexities these approaches often construct a model that capture relevant features of a data set the model can be used to make predictions about new data such as how well a patient will respond to a particular treatment or whether their cancer is likely to recur therefore the model is usually constructed using a large and
diverse data set which will then be applied to incoming cases to make predictions them in this video we're going to be learning about such methods that will explain how classification as well as feature selection works supervised machine learning takes place in data that has to be labeled and typically this is done by people in biomedical projects this could be clinical data or some other type of phenotypic data this label data is use the trainer model and the model will be tested on a test data set this data set will include examples of what is in
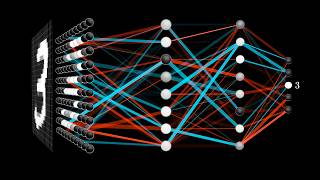
the training data the methods vary as do the principles that we'll be using to build the model but for now we can refer to them as a black box the method we'll essentially learn from the differences between various classes that are present in the training data set once the learning has been established you'll have a model or a template that can be applied to new datasets new data will have to be assigned with a class that is present in the training data set so if something doesn't fit such as the stars on the bottom right
of this screen model accuracy will be reduced the output of a classification algorithm includes predicted classes for each object or sample that's found in the data some algorithms such as random forests also evaluate how accurate and stable the prediction is evaluating which features if taken out will be the most significant for the accuracy of the prediction to illustrate this concept simply we can look at the classification of images we train the model by showing it images of cats and dogs and then testing that prediction on other variations of cat and dog images but instead of
cats and dogs we have gene expression to train the model we'll have to label the data by using phenotypic information such as cancer stage or subtype of risk factor and the training data we will have a special row that we will call class and it will contain labels for each of the samples genes and the machine learning language are going to be features and samples are called objects the test dataset will not have the classroom it will only contain genes sample names as well as levels of expression one such algorithm is called binary decision trees
their output looks similar to hierarchical clustering with each tree being created by thresholds and rules binary means the branch can either be yes or no if the expression of a genes is higher than X the classes will separate the algorithm will continue until it has effectively separated all the samples into some group if we use less genes in the training data set we will get different results because the data set can be dealt with differently also binary decision trees are initiated with random started so the genes selected in this procedure can change from time to
time here we see the cell line data separated into three groups luminal basil and Cloudland low with two genes random forests uses multiple instances of these decision trees that are applied to portions of the data one at a time after the analysis the tree predictions are analyzed the majority voting is accepted the algorithm is useful for more complex patterns where smoother borders between classes are needed it also gives an output of most significant features for classification linear discriminant analysis or Lda is a generalization of Fisher's linear discriminant which is a method that's used in statistics
pattern recognition and machine learning this method is used to find a linear combination of features they characterize or separate two or more classes of objects or events the resulting combination may be used as a classifier or for dimensionality reduction before later types of classification LD a is closely related to analysis of variance as well as regression analysis which also attempt to express one dependent variable as a linear combination of other features and measurements however analysis of variance uses categorical independent variables and the continuous dependent variable whereas discriminant alysus has continuous independent variables and a categorical
dependent variable ali-a is also closely related to principal component analysis which is also called PCA as well as factor regression analysis and that they both look for linear combinations of variables which best explain the data by contrast PCA does not take into account any of the differences in class in factor analysis builds two feature combinations based on differences rather than the similarities what's important to remember here is that Lda assumes that the data is normally distributed within each class so preparing the data before running the analysis is going to be an essential step wise Lda
makes it possible to automatically select those features in your data that are most useful or most relevant for the problem that you're working on this is a process called feature selection feature selection is different from dimensionality reduction although both methods seek to reduce the number of attributes in the dataset a dimensionality reduction method does so by creating new combinations of attributes feature selection methods include as well as exclude the attributes that are present in the data without actually changing them stepwise linear discriminant analysis is used to find a subset of the provided features that optimally
separate the classes that are inherent in the data in this procedure the discriminant model is built iteratively starting with an empty set of features ali-a classification is evaluated on each feature and the feature resulting in the highest accuracy is added to the optimal feature set other features are added to the set in the order of maximum improvement of accuracy until no significant improvement is provided by further features this is called a forward selection procedure where one adds features to the model one at a time at each step each feature that is not already in the
model is tested for inclusion the most significant of these features is added to the model so long as its p-values below some of the preset levels for instance this could be 0.05 this column fresh hold is set by the new Vogt parameter the input files are going to be the same as for the LDA pipeline for our expression data which contain expression levels for almost 7,000 genes we set this value to be zero point zero zero zero five in the results of the pipeline we found that it selected just nine genes but they provided a
perfect classification for the training set another important method we will talk about a support vector machines this algorithm differs from other classification methods because it is optimal to find the boundary of the class and uses a feature space transformation with a kernel function that makes separation more efficient for example a circular class border can be achieved by a straight hyperplane if the space was curved in such a way that the classes are cut by it with a decision surface the original SVM algorithm was invented by vladimir vapnik and alexei Shyvana kiss in 1963 vapnik suggested
a way to create nonlinear classifiers by applying the kernel trick to a maximum margin hyperplane the current standard incarnation of SVM using soft margins was proposed by Corina Cortez and Babinec in 1993 and published in 1995 this idea of support vector machines is to not the training data into a higher dimensional space feature via fee and construct a separating hyperplanes maximal margin there this trick helps to find efficient ways of separating classes while dealing with messy data that cannot be separated by linear boundaries as a result the transformation creates a new function for distance in
the new space an SVM model is a representation of the examples as points in space and their map so that the examples of the separate categories are divided by the clear gap this is why possible new examples have been going to be mapped into some space and predicted belong to a category based on which side of the gap they follow during this prediction stage the SVM algorithm will measure distance of a new object in the kernel space to the nearest representative in the training data SVM stands for support vector machine this is an efficient classifier
that can deal with complex separation between classes however it could have trouble working with too many features the input is trained and test data and the output is a predicted class with a guru CIL's for a prediction there are many considerations that should be made for supervised analysis first we should fit our classifier on a training set and predict the classes on the test set we also have to determine if it's possible to tune 7000 coefficients by 52 samples some of the algorithms do feature selection such as s WL VA and random forests however other
algorithms won't work if the number of features is in great access to the number of samples and then there's the curse of dimensionality these methods are commonly used in a variety of biomedical research as well as clinical projects for large datasets are beginning to be more and more available these include patient stratification disease classification and diagnostics identification of potential responders of therapies mechanisms of disease onset and progression and then on the form of biotech side there's biomarker discovery detection of toxicity of the in vitro stage the analysis of molecular mechanisms of drug action which is
also known as target discovery and then drug repurposing and repositioning









![Why Deep Learning Works Unreasonably Well [How Models Learn Part 3]](https://img.youtube.com/vi/qx7hirqgfuU/mqdefault.jpg)