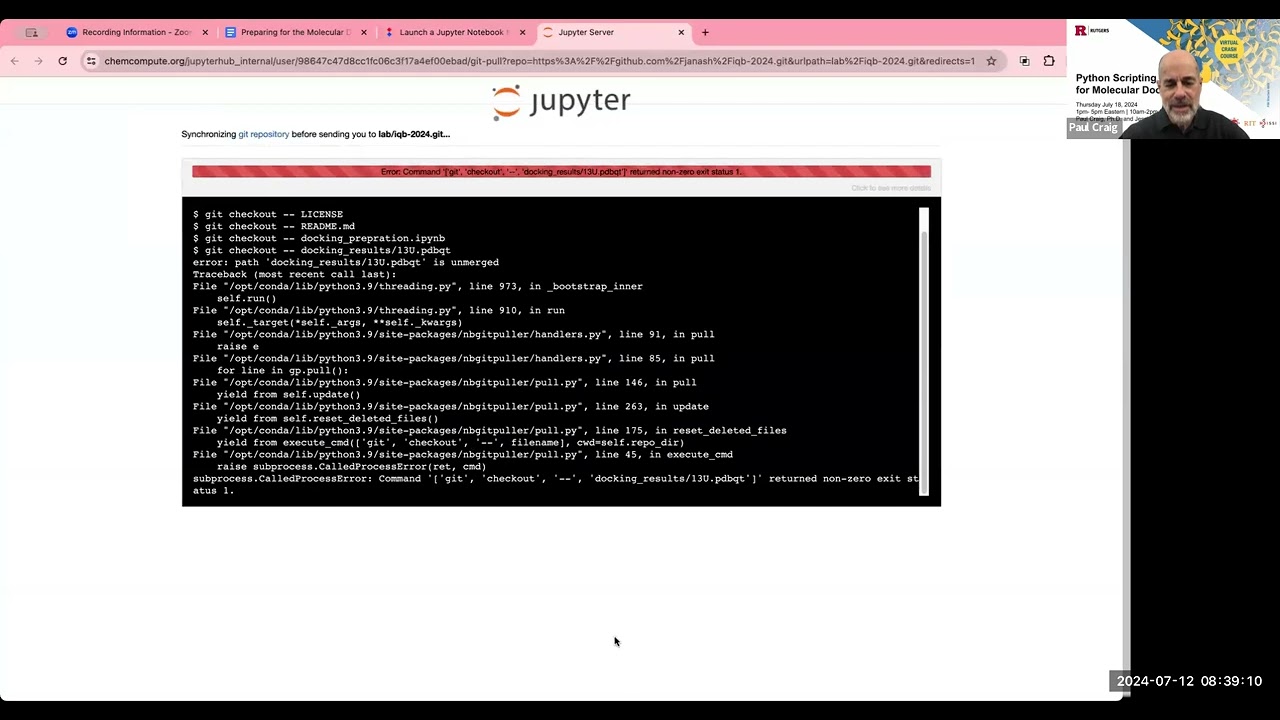
um and we again now that we have those input files to autod do Vena and what autod dova needed is a pdbqt file of our struct of our receptor structure our Lian structure and um to make that pdb file pdbqt file we had to adjust everything and add our hydrogen so now that we've done all that we can feed those structures into autod do Vena okay so this in this notebook we're going to actually be looking at running that docking calculation with autodoc Vina we're going to be visualizing the results from those docking calculations um using um a python Library called prolif which is for protein liant interface I can't I it's something like that I've got it later in the notebook okay so we're going to be learning how to um execute Li docking using autod do Vena we're going to be defining a lied box for docking so in order to dock something using autod do Vena we have to tell it what part of the protein we want to dock to then we're going to analyze and interpret our docking results using a library called prolith so um in this particular case when we're docking we already know where the docking is docking pocket is going to be we're going to base it on that experimentally determined structure we have a we have a structure with a Lian bound already so we know where we're trying to dot we did have a question and the a very good question in the Discord um during the break on sort of what if I don't know where I want to dock or how can I find alternate docking locations so there are there are um libraries out there that exist that can analyze protein structures and find potential binding Pockets um but we're not addressing those today we're just going to take a binding site that we know already so molecular docking it simulates the interaction between a small molecule and a protein and it predicts how the molecules fit together at the molecular level it's really commonly used in fields such as drug development um to design molecules to design to bind enzymes or proteins um during the stocking process um what's going to happen is we're going to tell autod Doven what our receptor is that's our protein we're going to tell it what our Li is and then Auto do Vena and then we're going to tell it where we want it to dock so we're going to Def find a binding pocket and then Auto Vina is going to take that Lian and it's going to try um various orientations and confirmations of the Lian within the proteins B binding pocket and for each pose that it tries it's going to assign that pose a score and that score is going to be based sort of on um some scoring function that's been predefined so those functions can be things like physics based so they can be based on force fields for those of you who have worked with force fields before for um they can be empirical and so um we're going to be using a scoring function called um Vina or vard Vena you can switch it out but autod do venina has three scoring functions that it can use two of them are empirical we're going to be using one of the empirical ones and there's even work that's done to make things like machine learning um scoring functions to predict binding Affinity I did include a link if you're interested just in information on scoring fun fun I did include a link to a recent review on scoring functions so that's what we're that's what we're going for and so the libraries that we're going to use for this notebook we're again going to be using OS for handling file paths and directories um we're going to be again using we're going to be using MD analysis again to measure things about our structure so we're going to be using MV analysis to help us toine that binding pocket for our Lian we're going to be using the Vina for um this is the autod do Vina python API to actually do our docking and then we're going to be using a library called prolith which is for protein liant interaction fingerprint that generates um an analysis of the different um types of uh interactions that are going on in that binding pocket once we've done once we've done the binding okay so before we dock I mentioned that we have to define the place on the protein structure that we want to dock to so we have to Define our actual docking lied box there are different ways that you could do this because we have a structure that has our that has our ideal Lian docked already we are going to use the location of that Lian to Define The Binding pocket that we're going to be docking to so if I were to look at my docking preparation I'll just remind you of the structure that we looked at we're going to Define our binding pocket by this Li that's already Bound in our starting structure before we isolated anything so we're going to be docking to this same location so we are going to load that structure you'll see here this is that pdb that we downloaded from the protein Data Bank we're going to um load that structure into to MD analysis and then we're going to select just the liance and then we can use functions in MD analysis to calculate where that liant is in order to Define our binding pocket so here we're um reading in that pdb we're selecting the liance and here we're going to use a um function in empty analysis so we'll give it Li MDA and that is the name of my wian selection and then I'm going to get the center of my binding pocket by just finding the center of geometry so in order to find to Define where we're doing the docking Vena needs two pieces of information it needs the location of the docking box center and then it needs us to actually tell us what the is and this first step we're taking the location where our Li is already bound that Center of geometry to be the center of what we're going to do uh consider our Bing pocket so again I did that just by putting it in MD analysis and I'm using an indd analysis function called Center of geometry and then I've got that printed here so I can print what that is and so this tells me the 3D coordinates of the center of my binding pocket for my docking okay so to after defining the pocket Center we're going to Define what the actual Lian box is and we're taking kind of a simple approach to defining this Lian box we're just going to take the minimum and maximum positions of our liin in each Direction and subtract them to get the sides of our L box and then we're just going to make it a little bit bigger by adding a five angstrom buffer around the edges um so you might choose to Define your binding po pocket differently um for our purposes we're just Define defining it in a very simple way to be the minim max of that already bound liant and then just adding a little bit of buffer to that so we'll add in here we had um Li box equals Li NBA um here is how we can get the positions of a particular atom group then we take the maximum we will want to subtract the minimum so we'll change we'll change this expression a little bit Li n da. positions Min so here we're taking the minimum um coordinate of the LI in each Direction and that's going to give us a partic so if I just ran that that's going to give me a particular box size so this tells me that my wigin docking box is nine units in the X Direction 12 in the y direction and eight in the Z Direction but I'm just going to make it a little bit bigger by just adding five to it so this just adds a little bit of a buffer um to each side of our box to make our um our binding box a little bit bigger so maybe if we're doing our docking calculations if we make it too small it's not going to be able to find you know there might be some binding that we might miss so we're just going to make it a little bit bigger okay so so far we have calcul used MD analysis to calculate our Center of geometry of our ligant and we've used MD analysis to calculate the size of our Lian Box by just considering the Min and Max coordinate of our liant and this last step here we're just converting those numpy these these that we've calculated those are numpy arrays autod do venina just wants them to be lists so we're just going to use this two list method to make sure that what the variables that we have to input to autod Vena are lists and not nump arrays so I'm going to fill in box to list it's not going to do anything about changing the numbers it's just going to change the data type um the data type of uh those variables okay so we're preparing for docking in order to dock there are various pieces of information that we needed we needed those pdbqt files that we created we also needed to Define where we are docking on our structure um there are different ways to do that we chose to take that Li that's already bound so now that we have those pieces of information we have our input files and we have our uh binding pocket we are now ready we're finally ready to do our actual docking calculation so this um in this cell we're going to get ready to do it so we're a little bit of file management here we're going to make yet another folder for just our docking results and we'll do this is the function that we've been using throughout the workshop to make directories and I'm going to make a directory called docking results and again I'll tell it if it exists that's okay okay so I'm making a directory called docking results and I am going to SC store my docking results in that directory I like to think keep things very separate so you know at each step here we've had different directories for different things we'll make that docking results directory just for the docking results and now that we have our directory let's actually do the dock so do the for for the docking we're going to import we're going to import our Vena library for the python API we do import V from from Vena import Vina and then we create our docking object and when we do that we give it a scoring function so here we're giving a scoring function name we are going to be using the Venus scoring function to do docking in this case so here's a little bit more about the scoring function fun that are available in auto. remember what the scoring function is for is it is the equation basically that is used to determine the interaction energy of our um Lian with our protein so what's going to happen when we do this docking is it's going to try a bunch of different poses and it's going to calculate a scoring function depending on it's going to calculate a value depending on the scoring function that we that we pick and we're going to pick vone I I'm going to say a little bit more about I'm gon to come back and say more about scoring functions while we are waiting for our calculation to run because we're actually going to see that the um the docking calculation is is a bit computer intensive so let's just execute this cell this is setting up our um docking object and it's giving telling what scoring function we want to use which is the vena scoring function after we do that we need to set up what our re receptor and our Li is so we need to um tell it what protein we're docking to and then we tell it what Li we are docking so initially we are just going to redock that ideal Lian um we're not g we're not going to dock the modified ones until we dock the the original one first so to do our docking calculation we'll want to set our receptor and here we're setting our receptor to be that pdb file that we um that we created created now we're going to set our liant and we had set earlier I've set my ligant 13u in a variable so initially we're going to um set our Lian to be uh we're setting it equal to that ideal liant and then we want to set our docking location so we're going to set that pocket where the pocket Center is as well as the pocket size so we calculated that already and that was in a variable called pocket Center and also in a variable called wian box so those were things that we calculated at the first part of this notebook so this is Computing some maps that are going to be used later okay and after we set that we actually perform the docking so here is the step that does the actual docking there are two I'm going to go ahead and execute this you can go ahead and run it there are two parameters for this docking calculation um there is a parameter called exhaustiveness and then number of poses and so we have our exhaustiveness for you're going to see that it takes a little bit to run here because it's actually doing a docking calculation and what it's doing is it's taking that Li and kind of fitting it um onto our receptor and doing and scoring it so you can see that it's still running so this exhaustiveness basically uh says sort of how many poses are going to be tried a higher number is better for getting you more docking results or better docking results the default value for exhaustiveness in autod do Vina is eight but they recommend in their docs for best docking results to use something like 32 so that is much higher than we want to set it right now because if we set it higher we're going to be waiting for several minutes but what that means is that the the poses that we see might not be that good so if you were doing a real cot docking calculation you would want to set this exhaustiveness to be higher we're also only writing out five poses you would probably want to add um write out significantly well me not you would want to write out more again if you were doing an actual docking uh calculation but again for the purposes of this Workshop just so we're not sitting here a very long time we've left these numbers relatively low um to say a little bit I said I was going to say something about scoring functions so we're using autod do Vena um we're using autod dovina and one thing about autod dovina is that um it has um three scoring functions if you want to use different scoring functions there are other open-source docking softwares that you can use so for example um one popular one that where the um scoring function is more customizable is one called smina and for SMA you can set a more um custom scoring function but ones that are available in autod dobina are the vena scoring function which is one that we're using this is an empirical scoring function where um we have different terms like hydrogen hydrophobic interactions and Stereos steric interactions um the scoring function was empirically derived from pitting data available in the pdbbind database um I've linked you to the original publication as well as the paper for the the next scoring function that's available as part of autodoc Vena called venardo and this is for Vena radi optimized um it's a it's to develop the scoring function by adjusting Atomic Atomic radi um of the original Vena function and then autodoc Vena also has a physics based scoring function functional available available called ad4 um this is the most computationally intensive scoring function um that's a available in autodoc Vina but it also we can't use it for our system because it requires the definition of a flexible receptor so we would have to go through some additional preparation if we wanted to use um this scoring function but these are ones that are available if you are using autod dobina and have prepared your files so I've talked through the scoring functions and now I'm checking so my docking is still going um I am seeing in the Discord that some people's docking is finished so hopefully mine will finish soon I will comment that my computer is running significantly slower with zoom than it usually does so I think that that's a an issue that we're encountering and yes I am running locally and it usually runs much faster so many people are saying in the uh um Discord that they are docked and ready to go so let's pause here for another minute I did mine in codespace and and and it finished about two minutes ago right yeah I think that it might have something to do with um sharing the screen for me yep I may stop sharing for just a moment to see if that helps apologies oh did you pray perhaps share should we keep waiting for mine or let's give it another minute and if not we'll switch over to my screen you can you can just tell you what to type okay tell me what to type yeah maybe for these computationally intensive portions we should have the Martha Stewart method where we've already got we've already got the cooked cake great idea you know what finished great okay I'm gonna share my screen again okay good yes and I am getting a very bad thunder storm where I live so I don't know if everyone's gonna start hearing Thunder but okay so my mine has finally finished docking and it's also like thank goodness I didn't choose something more computationally intensive right um so I've experienced this with autod Vena in the Jupiter notebook um it always gives you this little output here that says 100% depending on whether it feels like it or not it might also display the energies for you but luckily we can just access those in case it doesn't as in it hasn't now so um once your docking calculation is finished we're going to go ahead and write out your results so autod do Vena again it writes these pdbqt files and it's going to Output just the structure of those lians that it docked so for for the LI it found five poses did you did everyone never mind that was just a thunder thunder yes we heard it just a little bit not much so for um for those poses we have four we have four poses we're gonna write them out as a pdb QT file in our docking results F folder so I'm going to run that um it should run pretty quickly but now we have a file with all of those um poses saved so if you would like to see the energies that are calculated that were calculated for your poses we can see that by doing b.
energies so this is going to give us a numpy array where we have the energies that were calculated for our docking poses the First Column is the overall score for each pose so our top score here had negative 5. 22 and then these other columns correspond to um uh other types of energies in the docking score and so these are dependent on the um docking the scoring function that you use but for the Venus scoring function these correspond to the total energy uh the inter energy energ intra torsions and then um intra best pose um energies and I'm actually not quite sure what this last one is um but that's what each column in this is and um you might wish for your particular for particular docking calculation so I'll also comment that when it does these um docking these docking uh calculations there's a random component to it so each of us are going to have slightly different numbers that we see for these docking energies and we're each going to have also slightly different poses that we see um for our docking poses so um if we want to save these energies and return to them later I'm going to use pandas for that so pandas is another um library that we covered in a previous Workshop that's good for working with tabular data or CSV files and since I did that calculation and especially since it took me so long to do it I want to save um those energies in a file so I'm going to import pandas here I'm giving it those column the energy names um and if you are curious how I knew what those energies corresponded to I read the documentation for the software that's how I know what the different terms are I'm just um having a name for my columns and I'm going to create a data frame here where where I put in the energies and then I'll put in my columns so now I have a data frame with my energies and I'm just going to save that in a CSV file so that I can refer to it later if I'd like because I have the actual configuration of each pose saved in that pdbqt file I might want want to know the energies that correspond to each of those poses so I'm going to save that as a CSV in my docking results folder and I'll call it 13u energies. CSP okay and now if I go to my docking results folder I'm going to see that I have the poses for that first Lian as well as the energy is saved as a CSV file okay so now we want to actually visualize our docking results so after performing the docking simulation and saving the energy we want to look at the poses um so when you visualize results from molecular docking scientists are off often looking at both the 3D dock structure uh the 3D structure of the dock leg as well as a 2d representation called an interaction map and so an inter interaction map is something where we have our Li kind of um represented as a 2d structure and we have a map of The Binding site of the residues that are in The Binding site and then we have um annotations of the types of interactions that the LI is having with with residues in that binding site and so we are going to use a library called prolith in order to create both of those visualizations so to this is more fun with file formats autod Vena outputs a um pdbqt file but if we use prolith it wants our um it wants our small molecules saved as an SDF so we are again going to use Mo to take our pdb a pdbqt files and convert them to SDF and mo F fills in very very nicely to prolith that's one reason that we're using it but um you should note that if you want to um convert pdb to QT to SDF using Mo you need to make your pdbqt with moo originally so that's why we used moo in the last one so we're going to use a command another command line script from Mo called MK export and so we're exporting our pdbqt of our docking results to an SDF file so I'm going to run that and if I just take a peek again at my docking results folder I see that I have this SDF now of my uh Dock ligans and now I can do my analysis with prolith so I'm importing prolith as plf and then I'm importing MD analysis as well because I'm going to use MD analysis to load in my protein structure and again for um prolift that's another place where we want to use our structure with the hydrogens um because prolith does an analysis of the interaction it needs the hydrogens attached just like autod do Vena so we are loading in our protein file our protein structure with our hydrogens and then after that we are going to load in our structures so right now we have our structure in MD analysis next we need to load it into prolift so we are loading our Indie analysis structure of a protein into prolift with molecule from MDA and then we can just put in that SDF file from MO so that was in blocking results 13 u.