hi in this video I will talk about real-time quantitative PCR real-time quantitative PCR is one of the most important technique these days in biomedical research and also fundamental research so before going to the mechanism how real-time PCR works we should look at what are the utility of real-time qpcr Milton qpcr could be used for quantitative gene expression analysis copy number analysis differential gene expression analysis and also variety of diagnostic uses now real then qpcr could use two different chemistry's or two different reaction mechanisms one is a die based method which utilized cyber cream or one
is a pro based method which use TaqMan probes in this video i will talk about cyber bullying based detection mechanism now cyber green is a done so when it is not bound to any double-stranded DNA it won't flourish so in a solution we'll just have any double-stranded DNA cyber green would be there but it doesn't flow us now let's say that the same cyber green when it is bound with a double-stranded DNA it will start fluorescing so this is the whole concept behind cyber gray now just like a normal PCR reaction real-time PCR is a
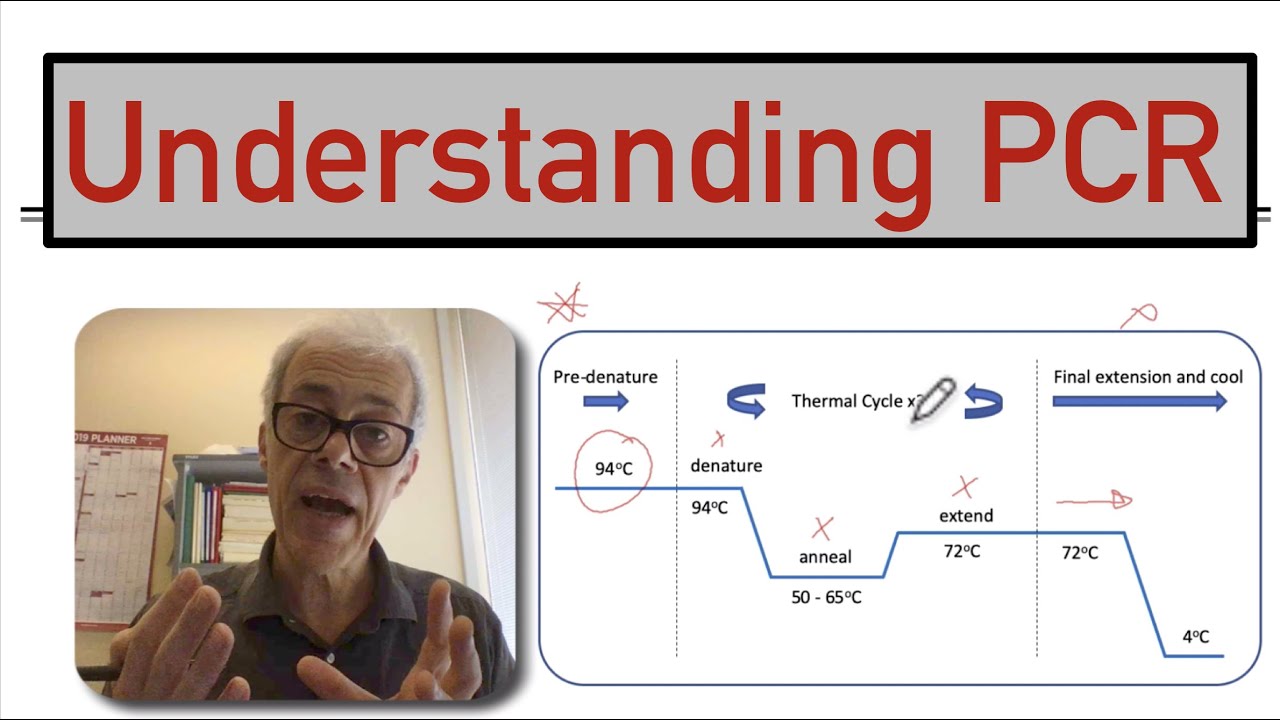
normal PCR reaction but the fundamental difference is we can monitor the progress in real time we can understand how the amplification process is happening so the first process the first step of this whole real-time model officia process is very similar to a PCR process which is a melting in the melting State the DNA strands would be melted as the temperature rises to 95 degree centigrade clean strands would be mailed it into two separate strands after that in a primer annealing step there would be a leading of the fragment and it would amplify it would actually
exchange the primers and in the extension state there will be another copy of DNA for now since cyber green is in the solution of these reaction mixture so cyber green can bind to any double-stranded DNA so you could bind to these double-stranded DNA and start tracing now after the end of another cycle there would be more DNA and there will be more cyber cream signals so it to PCR has a fluorescent detector so this fluorescent detector can dictate the signal coming from these DNA samples so over cycles there would be a huge increase in fluorescence
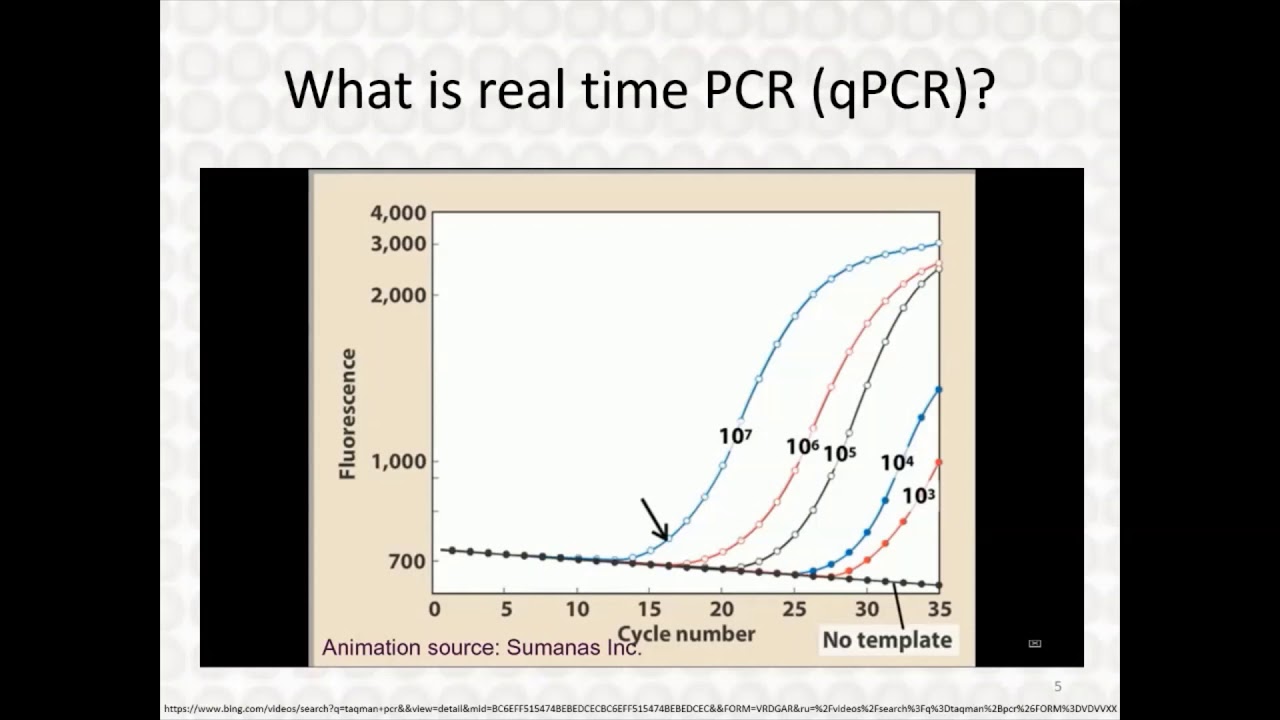
so for fast few cycles the fluorescence would be below our detection threshold but after a point the few loose fluorescence would reach the threshold and cross the threshold and grow linearly and after a time point it would saturate the point where the fluorescent rise above the base line is known as cycle threshold or CQ or CT well now here is a real-time amplification plot from real-life experiment where you can see where you can see here two amplification plots and here is the fluorescence baseline so for this and this is that for this particular curve family
of curves are for one gene and this is for another gene can see for this gene the amplification is near about 20 so the cycle threshold is near about 20 whereas before these gene clusters the cycle threshold is near about third so from that you can clearly understand that this chain is more abundant that's why it take less time less cycles to increase the fluorescence beyond the threshold level but this gene is pretty less amount that's why it takes more PCR cycles to detect the fluorescence coming from these amplified DNA and here is the quantitative
value that the Machine gives you now for on which parameters does the DLN PCR reactions actually the quality of the real will that we see a reaction is very for optimum reaction condition we need to do good primer designing most an amplification efficiency of real-time PCR reaction should be good there will be there should be less well - well variations and also the quality of the RNA and cDNA matters and last but not the least the my fitting efficiency is the most important part now how to understand that whatever amplification has happened in real name
quantitative PCR is specific or not let's say the lot of amplifications happened in the PCR cycle and these and these small products are known as amplicon now if we increase the temperature after the amplification what would happen they would melt and once they melt the cyber green won't be attached to the double-stranded DNA and there would be increase in the fluorescence levels so you can see here is the fluorescence level after amplification but one after amplification has the temperature rises so there are points where the fluorescence level sharply decreases so these are known as many
temperatures where the amplification melt and if you do a derivative of this curve you would get am a little peak so in one single male Pig means there is one single product but if you get multiple matrix that means there could be multiple different nonspecific products as well and if you run the gel so you would see multiple bands coming up but we should expect that our reaction is giving us a single product of the desired length so the length could be checked by running it into the tail and each time we run the reaction
for the same gene you should look at the melting temperature now when there is a contamination from a genomic DNA or when there is contribution from other sample or many other reason or there could be are nonspecific amplification there could be our target amplicon along with that there could be other applicants so these would give right rise to several steps in our middle peak so this kind of made it big is not okay and it is indicative of nonspecific amplification if this kind of milk peak appears then we cannot use the reaction for further analysis
so now how to get rid of that kind of nonspecific amplification so you know all genes all genes are made up of intron and exam so these part is the intro now once it forms a pre mRNA micron is still there but once the mRNA is processed these introns are spliced out so if you make a primer which is spending these boundaries then it would only amplify from the cDNA pool the target of it but doesn't amplify the genomic DNA content from from the genomic team so from the cDNA when we make C DNA from
this mRNA it won't have the intro so anything the primer would pick up would be in the boundary of this exon exon so it won't be picking up anything even if there is a genomic DNA contamination from this way we can reduce the chance of nonspecific amplification other possibility of nonspecific amplification is non optimal mainly non optimal annealing temperature of the primer so to mean a gradient PCR we can first check the proper annealing temperature of the primer and you can use that in our reaction to optimize our reaction properly so in this video I
didn't talk that much about the analysis of the real-time qPCR data but in the next video I will put that so if you like like a video give it a big thumbs up don't forget to Like share and subscribe thank you you