Hello welcome to the next lectures uh which will be about the gas chromat chromatography in the last lecture series we looked at chromatographic analysis of as such and in this lecture we are going to focus one of the two most important chromatographic techniques namely the gas chromatography in the LA in the next lecture so the last lecture of about the chromatographic um method we are going To look at liquid chromatography gas chromat uh in the gas chromatography lecture a lot of this is based on a legendary book by Harold mcneir basic gas chromatography um so
many figures come from this uh this book so if you are more interested in gas chromatography I definitely recommend you to have a at look a look at this book uh to to read more intensively about the uh about the gas chromatography Techniques um gas chromatography the as the name suggests is a chromatographic method where we use mobile phase as gas as a mobile phase uh the stationary phase in gas chromatography can either be a liquid or a solid and increasingly uh this is actually a liquid phase and nowadays used as the stationary uh face
face and the analytes that we are determining with with gas chromatography are mostly uh organic compounds and Volatile organic compounds uh however also analysis of inorganic gases for example is possible but the big big part of the samples are are analysis of organic compounds uh volatile organic compounds so why is gas chromatography such an important method firstly gas chromatography was actually the first of the two common chromatography methods that was um automated to the level that there were was um a widely available Commercial instrumentation so even though the first experiments that Michael twet um carried
out were actually liquid chromatography separation of the uh pigments liquid chromatography became commercially widely available only later than gas chromatography SO gas chromatography was already very popular in the 60s and 70s while liquid chromatography picked up pace uh after gas Chromatography SO gas chromatography historically is a very very efficient method of separating compounds um and uh it is it has numerous wide applications therefore uh it can separate very very many compounds simultaneously it is very efficient meaning that the uh Peak the num the theoretical plate numbers that can be achieved with gas chromatography are very
high so the peak um the plate numbers are somewhere around 100,000 Theoretical plates per um per analyst per compound so um this is um very very high and it's still much much higher than is achievable in the liquid chromatography uh though the number of compounds that can be determined simultaneously today is also similar for liquid chromatography especially after coupling with mass spectrometry but historically gas chromatography is more efficient and able to determine uh and separate many many more compounds so This is easily in the range of hundreds of compounds also with a suitable DET detector
and many detectors are available for gas chromatography the quantities of the compounds that can be determined are very low so if a suitable sensitive detector for the analy is chosen then uh determination of the compounds in the PBB range is easily possible but this strongly depends on the detector and on the compound that we Are wanting to quantify also with automation with the possibility to automatically inject uh samples to the gas chromatography systems hundreds of samples can be analyzed per day so this also means makes it suitable for wide scale analysis of environmental or pharmaceutical
pro pro samples or medical samples anything at all actually and uh it is also suitable for quantitative analysis Though usually internal standard methods need to be used to actually get nice quantitative results um however there are also limitations gas chromatography due to the fact that our mobile phase is a gas uh which means that the compound needs to uh in order to move with the mobile face through the column it needs to become volatile it needs to vaporize to the gas phase uh therefore the compounds that we can analyze have to be at least Somewhat
volatile so that means that their boiling points can't be too high uh which of course limits the the scope of the method for example automatically we understand that two two two polar compounds which have very um high boiling points can't be analyzed for examp sample proteins are completely out of the scope board I guess chomatography and canalysis and it's also not uh very nicely suited if any of the sample Components are not terally La terally labeled so because we use high temperatures in the in introducing the sample to the chromatographic system through the chromatographic process
and also in detection so if we have compounds that we are interested in and which are terally labeled then the then gas chromatography can be very problematic or if not absolutely not applicable um however there can be ways to overcome these limitations for Example one of them is deratization which is used to uh make compounds more volatile than they are uh and this can be done then by uh carrying out chemical reactions with the analytes that are themselves or Al not sufficiently volatile for the gas chromatographic analysis uh to give you some grasp how does
a chromatogram from AAS chromatographic analysis look like uh here is a chromatogram of a peppermint oil that has been chromatographed with Gas chromatography and the aim here has been to see the full richness of the aromatic compounds and especially Aroma compounds in the gas with the gas chromatographic analysis and you can see that this specific chromatogram has been um obtained by a chromatographic method of 45 minutes and we have here actually very many Peaks um from the aroma aroma compounds 29 have been labeled uh but there are actually many more other compounds which are then
not as Associated with the aroma and you can also actually see from here how how efficient gas chromatography is the Peaks are super narrow it's maybe hard from here to um evaluate how narrow exactly but peaks in the second scale are completely normal in the gas chromatographic analysis uh going a bit in front of the topic saying that in the normal HBC the Peak widths are usually about half a minute or even longer and in um high performance or ultra high Performance liquid chromatography they could be in the scale of of 10 seconds or something
like that so the gas chromatography Peaks can be extremely narrow compared to the liquid chromatographic Peaks and um actually it would be a good um to think for a second why are then gas chromatography uh Peaks narrower what are the uh what are the processes when we think about the one damer equation that are so Much more efficient in the gas chromatography compared to the liquid chromatography and we can come back to this um in later discussions with you but think about it so uh the question now is what are the basis of Separation in
the gas chromatography how are the different compounds separated so uh the separation similarly to all other chromatographic methods is or almost all chromatographic methods is Based on the interactions between the stationary phase and the analy or then the mobile phase and the anite and uh in here the interactions can occur between the analy and the stationary phase and uh this um the this these interactions are largely driven by the volatility of the compounds so compounds which are less volatile are stronger interacting with the stationary phase uh however stationary phase chemistry so the polarity of the
stationary phase can add Additional interactions to uh the separation mechanism traditionally the stationary phases are not very polar and therefore the interactions between the stationary phase um and the analytes is driven by the volatility of the compounds so the compounds which are more volatile are more of their time in the gas phas so they are easily Vol volatilized and they move faster through the column while compounds which are less volatile are spending more time in The stationary phase and therefore move slower through the gas chromatography columns and so a rule of thumb is that compounds
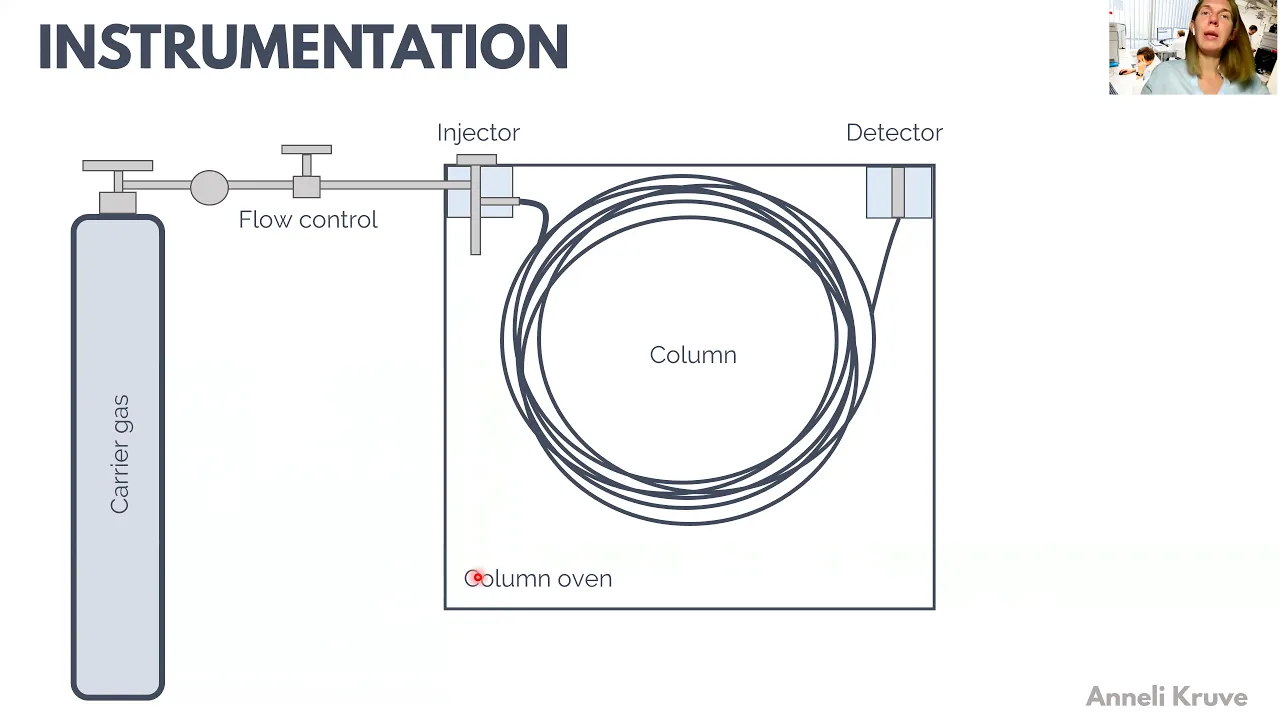
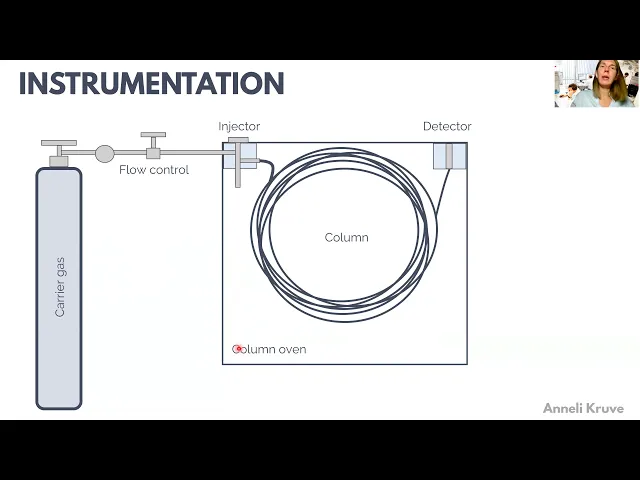
which are more volatile are less interacting with the stationary pH so how to practically carry out gas chromatography and for this we need an apparatus to uh carry out the chromatography and and everything starts with the mobile phase with the carrier gas uh which is directed into the chromatographic system and very Important PL here is the flow control which means that uh we have to keep the flow constant uh at all times of doing the chromatographic analysis and this is extremely important to uh achieve achieve reproducibility of our chromatographic systems also we usually want to
purify and dry the carrier gas uh to make our columns live longer and uh Not to cause any unwanted reactions between our samples and gas um and Carrier gas as well as the stationary phas in the column uh we used to bring the sample to the uh column with an injector which can be heated uh with a with a heater and there is usually a possibility to do to split the flow after the injector so that only part of the sample actually goes to the column and part of it can go to the uh can
go to the waist then we also have a column here The columns which are to nowadays usually capillary columns are very long so that's why you have here this long uh L um column with with very many uh turns and the column is placed in a column oven which is important to regulate the temperature uh of the separation because the compounds need to uh move from the stationary phase to the gas phase to move through the column it's important that the vapor pressure of the compounds would be constant uh from one run to Another and
therefore we need to keep the column temperature very precisely regulatable and therefore we need to have a column oven uh where our column is placed and where our separation chromatographic separation is carried out and finally to register the compounds once they elude from our column um we have a detector we can have here numberous different and then we probably have here also a computer system that actually registers our Chromatographic Peaks so the first uh important part is the mobile phas and as this is gas we also often call it a carrier gas uh it's it
job is to carry the sample the analy through the column and at the same time it should itself not really re um anyhow interact with the stationary phase so it should be inert uh and it should be also inert in this sense that it should not react with the sample and then also not with the Stationary phase so from the inertia point of view nitrogen is a very good uh carrier gas as well as helium and rarely though but historically hydrogen has also been used as a carrier gas Today hydrogen is not uh very much
used even though it sometimes can be cheaper than helium for example in some countries uh but it is a lot more dangerous to actually operate so hydrogen is usually uh avoided if possible and primarily Nitrogen and helium are used um it is important that independent of the exact choice of the carrier gas it is as pure as possible and especially important is it its freedom from oxidizing agents so oxygen and water and oxygen and water are very very important to avoid any possible reactions between the carrier gas and the stationary phase so if there are
residues of oxygen for example um it is possible that the carrier gas or Actually the oxygen in this carrier gas would react with the safe stary phase material would oxidize it and therefore the stationary phase would age and it its chemical properties would change during this oxidation so chromatography would not become any more reproducible and the retention times that have been observed on one day would not reproduce to another day and it's very likely that Peak start tailing because these oxid oxidized stationary phases may have very Different ret ret mechanisms than the original stationary phase
So to avoid this kind of uh problems with stationary phase it's very important to uh use as pure as dry and as oxygen-free carrier gas as possible and usually a number of different um purification um columns are used before the carrier gases directed to the real uh column where then the hydro the water is absor absorbed by the by these purification columns and also other Residues can should be removed also for example in the gas um in the on top of the gas cylinders where the flu Regulators are it's very important that there wouldn't be
too many alkanes used there uh in the oiling of the systems to cause any termination of compounds that could um also be detected with the detector so freedom from these compounds is also very important uh there is a special system in denoting the purity of the carrier Gas and it's usually denoted something like 5.0 or 4.0 and this five indicates the number of Nines in the percentage of the Purity uh and then the zero means that what after which after how many nines come this the first zero so if we say that the period is
5.0 then this means that the Purity is at least 99.999 uh percentage 6.0 would be that the pur is better than 99 point and then 49 uh Percentage uh the terrier cast also needs to be compatible with the detector uh and for example the limitation here is that for Mass spectrometric detector helium is needed and nitrogen is not suitable because nitrogen itself is also and ionizing in the mass spectrometry and it causes high background therefore uh it's also important to regulate the flow of the gas um that carrier gas as as precisely as possible and
this is uh Because we use usually use uh retention times for identification purposes and this means that retention times need to be as reproducible as possible but if um the carrier gas flow changes by let's say 10% then the retention times will also shift by about 10% and if a chromatograph then our sample at one flow rate but then the standard at another flow rate where where the uh flow rate was let's say 10 point 10 10% low um lower than all retention times Are roughly 10% longer which means that um then we would not
be able to actually accurately identify which compounds uh belong to which peaks in our sample so this reproducibility of the uh flow rate of the mobile place is extremely important in gas chromatography now uh the sample is injected to this carrier gas flow and it is important that the sample is very rapidly brought to the gas space so that it can be brought with as narrow band as possible From the injector to the column uh to keep the Peaks as narrow as possible and to avoid any kind of peak broadening so it has to happen
very rapidly and and actually two things have to happen very rapidly here firstly the compound the sample needs to be volatilized as rapidly as possible and secondly the sample needs to be carried to the uh to the column as rapidly as possible and therefore the injectors are Systems which are usually uh heated to a high temperature usually the temperature is is uh about 50° higher than is the boiling point of the sample to bring as fast as possible all of the sample components to the gas phase and then rapidly direct them to the column uh
it is important that uh this is sufficiently high so everything is vaporized but it's also important that um the sample would not decompose in the injection during the injection so this This from here comes the limitation if some of the compounds are becoming are being um uh are being to label temperature label so that means that they would decompose at the injection injector temperature While others maybe aren't even volatile yet this means that probably this kind of analysis is not sufficient suitable for the sample so all of the compounds of Interest have to be um
Sufficiently temperature label so that we would be able to get all of them into the column um so that they wouldn't decompose or anything uh when we have liquid samples then we usually inject a microl around one L microl can be less can be slightly more uh especially when we use capillary columns when we use uh when we use packed columns then these volumes can be even up to 10 microl lers and the injection is usually done manually with A micro syringe where manually the analyst can take the the amount of sample that is needed
to be injected or it can be done with an AO sampler and increasingly the AO sampler system is used a Samplers are of course more reproducible uh the injection volumes um but it's all Al the advantage that analysis can be carried also out of working hours which means that um the samples can be run also overnight which is of course a big Advantage when we Talk about large Cort analysis or or just very many sample analysis uh that need to be carried out it's also possible of course to analyze gases and for this gas typ
syringes for example can be used to take the sample and gas injections are also possible both manual and with an AO samp and for the injection systems there are very many different actual modes the most common ones are split split less and on column Injection and we will look more into the split injection which is kind of a traditional way to analyze samples in the capillary column U analysis uh but before we get to this uh just um maybe a caution PL here even though the liquids and gases both can be analyzed um in case
of liquid samples uh all of the compounds actually need to be volatile um which means that the boiling point needs to be sufficiently low but what is especially problematic is when We have mixtures of gases and liquids and one of these cases can be solve with something which is called a head space analysis so if the analy that we are wanting to analyze are sufficiently volatile then we can use the analysis from the gas space even though our sample itself might be a liquid so schematically it's it is represented here like this that the sample
consists of three different molecules for example but the Unaligned molecules the orange dots here are so volatile that if we put this sample even though a liquid into uh one a closed cap while uh then and wait for a sufficient time for the gas phas and the liquid base to equilibrate then with some time a sufficient part of the anal like molecules actually goes to gas phas and in this uh case we could actually sample with the syringe these unaligned molecules oranging dots from the gas Space so that we would not even need to inject
the rest of the compound pounds or the rest of the Matrix um from the liquid phase and it can be in this case when the compounds of Interest are sufficiently volatile that we actually take a gas sample here from the head space so with head space we mean the gas phas that is on top of the liquid uh in a closed while or that we use an additional solid phase uh to collect the sample and then get it out from the um From the from the while but the idea is that we still sample from
the gas pH we will look at this uh headspace with a fiber actually also in the sample preparation uh lecture that is coming in the quite end of the of the cost but the main idea here is that if the compounds are volatile then we can also make analys just directly from the gas phas on on top of the liquid phas and this is for example one um solution to analysis if our samples contain water so Generally water simil is um at higher temperature it is a oxidizing agent and therefore we our sample should be
as dry as possible so we can't inject water samples which is kind of a problem because very many samples that we might be interested in are can be in water or are natural in water so there are two possibilities here uh we could either do an solvent exchange with us during our sample preparation we could use a liquid liquid extraction or solid phase Extraction and we will look into the mechanism in the sample preparation lecture uh later uh or we could if our analy is sufficiently volatile we can try to use this kind of headspace
analysis the heads space analysis is for example very suitable if we have large water content rtion samples and we want to determine for example ethanol or other similar small um short chain organic molecules uh for example ethanol from blood can be done like that or Ethanol from the uh windshield liquid uh even though and this can be done without changing the solvent so sample preparation can be very very minimal uh just putting the sample into this V and analyzing the gas Ray from the from the top of the sample however other samples may need to
be uh dissolved or diluted with a suitable organic solvent that itself also has a sufficiently low boiling point so now about the injecting Injection with the split uh mode so split mode is a very often used in the capillary gas chromatography so cases where the columns are capillary columns and the reason here is that um when we want to when we inject our sample we want to get the sample to the column as a very narrow band to not make already when injecting the Peaks very broad so this means that um we should avoid very
large volumes of gas to the um entering to the column and when we do the math uh With the idle gas equation then we see that injection of one micr of benzene would result in 600 microliters of gas which means that that is quite a lot and actually it would take tens of seconds to get such a volume of gas to the capillary column and it would essentially uh overload the column which means that our Peak would become instantly broad just because the volume injected to the column would be too large and we want to
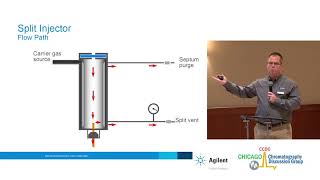
avoid this peak Broadening due to the injection of the large volume we can't really very much decrease the the uh sample size that we inject one micr is about in the range where we can still somewhat reproducibly uh take the sample volume so we can't narrow this down 100 times it would be essentially impossible so therefore a a smart solution called split injection has been uh designed and this is a a as injection type where we Use a system where majority of the sample is not directed to the column and let's see this here on
the example of the injector here so the sample is injected from here with a with a syringe so the needle purses first the septum an inner septum that should not U somehow cause any compounds to enter the column it should be as inert as possible and it also avoids the carrier gas to leave from the system by in this direction so it is only uh permittable With a syringe and with a syringe needle from from this side so the needle enters to this into this injector and in here the important PL part is the gas
glass line which is In N and should not react with the sample or should not uh be absorbing any anal light or anything like that and this is heated so this the syringe the sample is carried in here the injection is made by pulling pushing down the plunger the sample enters here and the In the GL on the surface of the gas glass liner the sample is rapidly uh volatilized the carrier gas is flowing through the injector and is carrying this um this Vapor rapidly vaporized sample downwards and now there are two possible exits for
the sample uh one which leads to the capillary column and another one which is called the split went or uh is or can be thought also as um essentially waste line so the sample that is Directed for analysis leaves to the capillary column but the rest is then directed to the waste essentially and the split ratios that can be used are somewhere the smallest can be somewhere to 1 to 10 so this means that one part goes to the column and 10 goes to the waist but most usually are somewhere between 1 to 50 to
1 to 100 so about 1 to 2% of the sample actually uh goes to the capillary column and this split uh injection is very important to keep the Columns uh the peaks in the column narrow and therefore enables us to get high resolution and it's also therefore suitable for analysis of Fairly concentrated samples so we don't overwhelm our column or overload our column and also dirty samples can can be uh analyzed because all this dirt is not reaching column majority of this of this is um going to the waist and in some cases even if
the glass liner additionally is uh filled with some kind Of inert material such as glass wool uh very dirty samples can be injected so that the glass wool is uh just absorbing the major part of the dirt and then uh only the well vaporized part is actually injected to the color but the problem is that we are when we are dealing with compounds which are really really close to the detection limits then we would not really want to waste a lot of our sample and direct it to the waste but we would actually like To
get it to the column and being detected after the column and in these cases splitless or on column injections are very useful um there are also other techniques such as cold down color and and others which are more which are um also used for um for solving this additional problems so split injection is kind of standard in the capillary column analysis but uh for other samples there could be need for for other other Injection modes as well now when we have our sample vaporized and directed to the column then we should also look at what
is this column then or or which kind of columns we have in gas chromatography and largely divided we have capillary columns and we have packed columns in case of capillary columns the stationary face is covered on the inner wall of a very narrow capillaries so these capillaries are about something like 0.25 mm in the internal diameter and the walls are car are then functionalized with the stationary phase which is a liquid and the interaction of the compound is uh of the analyes then between the iner gas flowing in the tube and between the stationary phase
on the walls of this capillaries and um kind of more traditional are packed column where round particles have been first functionalized with the stationary face So then there is an inert particle here with a blue dark blue color and then the actual stationary phase that causes retentions of the retention of the compound so this liquid phase is uh covered on the as a layer on the stationary face and then the the stationary phase is used to pack this column so it's tightly packed with the with the particles that are carrying the stationary face so for
the capillary columns which Are also called wall coated open tubular columns so C columns uh usually silica capillaries are used uh which are sufficiently nerd and temperature stable and to be actually um placeable to the column ovens and uh usable during the chromatographic processes the good part of the capillary columns is that the resistance to the flow isow is low which means that the columns can be made Fairly long uh usual columns are somewhere between 50 m to 30 M and but also very long columns have been made um so somewhere like 100 meters are
all are commercially available there have also been interesting trials to make very very long columns but uh in case of very long columns the efficiency starts decreasing again so it has been found that uh for a pretty standard sample uh roughly a 30m column is uh quite good choice as a first option another important parameter in The capillary columns is how W how thick is the stationary face that is actually Co um covering the inner wall of the capillary column and there are two competing processes firstly uh when the when the layer becomes too thick
then the then the res then the diffuse Fusion be in the stationary phase becomes too uh slow so in the one equation our C term which uh is how efficient is the Diffusion of the unal lighted to the stationary mobile face border becomes too inefficient and therefore B Peaks become broad so we don't want to have a too thick layer on the walls of the column however when we make it too thin then the The Columns are not able to take up two large quantities of the sample so their capacity becomes very um very low
which means that we can inject only very small sample quantities and only very low concentration samples so This is also something that we don't really want uh therefore kind of a standard is to have 0.25 micrometer thickness of the uh stationary phase on the inner wall of the column um however in some cases where we need to improve the capacity for example we have very volatile compounds or we need to inject very uh larger sample quantities then we can also have larger um thicknesses of the stationary phase on The column and uh the capillary columns
are very good because they are very efficient in separation of s of complex samples due to their ability to keep the Peaks very narrow and keep the efficiency very high and from the efficiency point of view the theoretical plate numbers of 100,000 or above are are completely normal to be achieved with um out of the box capillary columns now let let dig into the Stationary phase uh that is then causing the retention of the compounds um important part of the interaction between the anite and the stationary phase is polarity so let's revise what kind of
interactions we may actually have between the analy and the stationary face uh to remind when we talk about polarity then in case of pure liquids we would mean the dip moment of this liquid uh so this is when we just have One liquid in in a beaker or in a flask and we would think oh how polar this this liquid is then we would look up what is the dipol moment of these molecules and we would be able to say okay this solution is this uh liquid is more polar than the other liquid and this
uh dipo moment directly influences also the boiling point so the uh higher is the dipole moment the higher is also the boiling point more polar molecules are having higher Boiling points but in here in gas chromatography with polarity we are actually meaning something which is um it is a combination of the uh polarity of the solute soolly and the polarity of the stationary phas so we have thinking about the interactions of the unlight and the stationary face and Bly said we have Wonder W forces that are causing this interactions and we have hydrogen bonding uh
both of these contribute to The interactions and from the Wonder forces we have also three different types of interactions that we can have we can have dispers or London forces which are induced typle induced typle interactions and these are are occurring for all compounds independent of any um of the presence of any Polar Polar Bonds in these molecules so any dipoles and this means that if you have a stationary phase and if we have a solute that don't have any dipoles this is uh still Present and this uh is causing the compounds to be retained
still and this is also very nicely correlated with the boiling point of the compounds now if one of the compounds either the station or the salute has dipoles so there are some polar um bonds that cause a dipo then we can also have dipole induced dipole interactions and this now is something that comes in addition to the boiling point so this means that uh if the dipole induc dipole Interactions can be present it might not be Pur L the boiling point that is determining the illusion order of the analy and if both the anite and
the stationary phas have some uh ballar bonds so we can have diple diple interactions then these can also additionally increase the inter um the interactions between the analytes and uh stationary phase and cause the polar compounds to rain stronger than could be just um this SED based on the boiling Point however we have to keep in mind that the boiling point is also actually capturing uh the dipole dipole interactions between the analyzed molecules so uh if we have any kind of um bowar interactions the boiling point is a Rough Guide but uh almost never we
don't see exactly polar um exactly boiling point um increasing uh retention times unless we have uh homolog Series so if we have homolog Series where the functionality Is exactly the same the polar functionalities are exactly the same from analy to analyte the only change is the alkal chain length then we see extremely nice correlation between the retention times and the boiling points of these compounds because the only thing that is changing is then the dispersion forces between the alkal chain of the homolog series unalike molecules and the stationary phases uh in addition to the Wonder
Val Forces there can also be hydrogen bonding playing role and this is especially true for the uh polar stationary phases that we will have a look at and um or actually the most polar stationary phases and uh the problem of hydrogen bonding compounds can also occur so so sometimes the hydro compounds that can give hydrogen bonds uh are tailing in their on the chromatograms and this can be caused by The Unwanted absorption to the Stationary phase or to the uh slightly exposed um capillary walls or to the liner in the injection system and therefore functionalization
of possible sanal groups so mostly the um other part of the hydrogen bonding that the analy is giving the hydrogen bond with is a cenal group either then on the glass in the glass liner in the injector or on the on the in the column material on the uh silica material um so these penol groups Can be functionalized to make them more inert and avoid this tailing for the more polar compounds so now we actually have uh the stationary phases which are of very different polarity so there are some which are arbitr say to be
polar and some which are said to be very nonpolar and um the the polarity of them is um the polarity range between these two um between these two ends um can be Measured and comp appeared and there are tables where you can look up which stationary phase is how polar compared to another stationary phase however um it is uh still sometimes um not only possible to look at the tables with these different polarities but also to actually try out uh because of specific interactions can that can occur between specific compounds with functionalities however uh there
are some general guidelines that all Stationary phases uh should be um fulfilling to be usable in the gas chromatography so most of the stationary phases today are liquids um and this is also why a kind of um term gas liquid chromatography can be used or has been used um to denote that the actually the stationary phas is liquid uh this is was especially useful term at the times were also um really um solid particles were also used as the stationary phas in the in the packed Columns uh the liquids however need to be low vapor
pressure liquids which means that they would not themselves um vaporize and get and escape the column so we would need them to be very stable in the column and not to come out come out from the column um and they would also need to be terminally stable so different stationary faes have different temperature limits and uh it is very important that the St that the temperature limit would be much above The cellum temperature that we actually want to use to carry out our chromatography and the low vapor pressure and teral stability is also important to
uh ruce something which is called column bleed and column bleed is the stationary phase uh SL in time escaping from the column so just being kind of washed out from the column and making the stationary face constantly thinner in the column and also causing background in the detectors very often Uh when we are using generic detectors that are able to detect uh very many compounds so we want to keep this bleed so this leaving of the stationary phrase from the column as minimal as possible so therefore uh the stationary phase the liquid as a stationary
phase should be low volatility and it should be also temperature stable it's also good when it has a low viscosity so that the mass transfer uh in The uh would be as fast as possible so that we do would didn't cause any additional Peak broadening and um it's also important that it would interact with the analyte so either due to the dispersive um interactions or due to the uh dipole induced typle or diple dipo interactions uh kind of classical stationary face with um the with the least polarity is sine uh this is a high molecular
weight Alen with a somewhat Branch Branch chain and um this is termined as the least polar uh stationary phase and in the stationary uh stationary phase um tables where the polari have to compare the polarities of the stationary phases this is kind of the zero reference point with the lowest possible polarity and this comes from the fact that there are no um no functional groups that could give any other interactions than the D then the induced typle induced typle Interactions um the problem with sine however is that it has a very low upper usage limit
so this is only 125 degrees in celsius which means that um if we want to increase the temperatures then this is highly problematic and therefore uh this is now not very much used and actually um silicon polymers so polysiloxanes are highly used and the least polar polyan which is called also ob1 uh is a polyoxin which has mthal groups attached here uh so mthal groups Are also able to give uh induc typle in typle interactions and the least polar is uh the column where only mesal groups are attached to the polyu now in order to
increase the polarity of the polyoxy stationary faces also fenel groups or Sano groups can be attached instead of the methal groups and the ventilation percentage can be somewhere between 5% which is still relatively less least polar stationary phase to about 50% which is then a Medium polarity um stationary phase but in the um silic silicon polmer stationary pH faes it's a fairly polar uh stationary phas and fenel group grou are making the stationary more polar because they have uh the induced typ balls are stronger in the Benzene rings than they are for the methyl groups
so if our unalike molecules are having dipoles so they have polar groups then the induced dipo dipo interactions are stronger with The fenel groups than they are with the mthal groups and therefore um even though we aren't introducing ourselves to this stationary faes any polar bom they are acting as more polar stationary phases due to the fact that the induced dipo dipo interactions are stronger with the fenel groups than they are with the Mel groups um because these are poers they are also more stable to the temperature and um they the stability can be up
to Three 350° or maybe even 375° uh so this is the temperature that can be used to um chromatograph which is a fair fair temperature range and mostly most of this uh chromatographic separations are done in this region however sometimes there is a need to get have even higher polarity to obtain even better selectivity and for these cases the most polar stationary phase is a carox uh car or carox 200 M Uh 220m sorry uh 20m refers to the uh molar mass of the carox and uh carox is essentially a polyethylene ccol which is uh
polymerized and then used to cover the inner wall of the capillary column uh it is a bit less stable than the Silicon paste volumer so up to 250° uh but it gives additional selectivity due to its additional polar interaction so here we actually have this um Co bond which is polar and also The oxygen is able to give hydrogen bond so this um gives additional selectivity here compared to the um mulated silicon polymers so let's us let us look what kind of selectivity changes can the stationary phase cause um so here are two chromatograms of
the same compounds so the heptin tetr hydrop Forin two um two ban banon as well as um n propanol and they have boiling points in the range of 64 Dees to 98 degrees in celsius and um these compounds have been Uh chromatographed with um polar station face here above and with a less polar uh stationary face here so the upper one is with the carox so the most polar stationary face and we see that even though uh heptane has very high uh boiling point in this in this uh among these compounds it actually loots as
the first one from The Polar com column and this is a um understandable as this uh compound Heptane doesn't really give any uh polar interactions with the stationary phase however uh propanol which also gives hydrogen bonding is the last uting compound and now interestingly we can here also compare the elution order of the compounds from The Polar stationary face to the um non polar stationary phasee and we can see that actually here the re order of the compound solution is completely reversed and we also see that even for the nonpolar stationary phase It is not
directly linear to the um to the boiling points of the compound so there are so the retention is affected by the boiling point of the compound but it's not uh a straightforward uh correlation now in order to actually carry out the chromatography with the gas chromatograph we need to achieve conditions where all of the compounds that we are interested in and that are or actually all of the compounds in our Sample would be uh able to pass through the column and at the same time should also be separated so we would need the column temperature
to be sufficiently high that all of the compounds actually pass through the column so meaning that they would not only be stuck in the stationary phase but they would actually partion between the stationary phas and the mobile phas and would actually slowly pass through this uh through our Column at the same time what we also don't want is that all of our compounds would be in the gas phas so we don't want that the column temperature is so high that the compounds are all in the mobile phase or at least many of them would be
all completely in the mobile pH because what happens then is that then they are just in the mobile phas and they all pass the column very very fast so we wouldn't get any separation of the compounds here Either uh usually we can achieve this by having a column temperature that is much lower than the boiling point of the of the sample and this is indeed uh due to the fact that we need to have our compounds portioning between the stationary phase and the mobile phase pH we don't want them to be completely either in the
uh mobile phase or in the stationary phase if the temperature is too high they are always in the mobile phase if it's too Low then they are stuck in the stationary phase and don't move through the column at all and finding this temperature is the critical part in the separation of the compounds in the gas chromat chromatography and can be very very challenging so look let's look at an example of a chromatography of uh uh n parapines which are a homo series and here above on the above chromatogram uh these compounds have been Chromatographed at
150° Celsius and what we can see is that the lower paraffin with the shorter chain length uh they are all kind of close here together and some of them aren't even really separated uh while after 95 minutes which is one more than one and a half hours uh the largest paraffin that has looted from the column is C15 so we could easily think that this is the last Paraffine um so we could stop here chromatographing and saying okay that was it but actually there were also other longer chain parapines here um and what what we
also see as a problem here here on the upper chromatogram is that the peak become Peaks become broader uh as they elud later so the peak that eludes the c-15 peak that eludes here at 1990 minutes is almost 5 minutes broad while the Peaks that eluded here in the very beginning Were like minute or less broad so this chromatography system where we use one temperature and they call this the isothermal Run um is not really very sufficient for this kind of a sample where the boiling point of the S of the compounds range in a
very very wide range and and to solve this there has been uh created something which is called a temperature program and this is the case where we rise gradually the temperature of the column as we Chromatograph so we start by one fixed temperature and then we increase the temperature and for the an example of the power of this temperature program we can see here from the same paraffin separation the initial temperature was 5 50° and it was Rose to 250° by 8 degrees in a minute so we can see here two things firstly uh because
we started with a lower temperature then was used here in the isothermal Separation the first Peaks are also nicely separated we see C6 separated from C7 because the boiling point for both of them is uh higher than the re initial uh 50° that was used as a as a very first starting point of this um temperature program and we see a nice separation of all of these compounds but what we also see is that there are actually many more compounds than we saw in the isothermal uh separation we can see here also C20 and C21
and in spite of the fact that they have longer attention times they are not necessarily broader Peaks so we get a significantly better signal to noise ratio and therefore also the LOD for the c19 for example in here so we see that the peak is much much narrower and much sharper and the peak is much better to separate uh from the Baseline in case of this uh temperature program chromatography and so therefore we see that with a temperature program which is Increasing the temperature of um of the column gradually during the chromatographic Run we avoid
B broadening with the longer retention times we also get the analysis done much faster so we see that here the last Peak is eluding at 36 minutes while here it was uh much more than 95 because at 95 we only got um c19 eluding from the column and and c21 we didn't see at all was this time as well as because the Peaks are get narrower we get better Signal to noise ratios and therefore lower LOD values so temperature programs have uh significant advantages for the uh for the chromatographing of samples where the boiling points
of the analytes have a wide range temperature programs are therefore ideal for uh screening of the sample when we don't know generally how many Peaks we are expecting or what kind of retention times uh we are generally Expecting to have for our samples so therefore in these situations uh temperature programs are ideal and temperature programs don't very vastly um applied even if the final aim is to get an ISO isot temperature uh isothermal separation in the end uh doing a first screening with the temperature program is is um very good to get a good overview
of the uh sample and at what what times what temperatures the compounds are eluting From the column it's also very good because column cleaning is kind of built in um the problem can be with isothermal separation that um Matrix components which have low volatility gets uh are not eluded from the column with the isothermal uh separation and therefore uh can kind of build up in the column and um cause column properties to change or to increase Baseline so for temperature programs because the temperature goes up in the during The during the temperature program these High
um low volatility Matrix components are also more likely to get cleaned out from the columns so this is good for preserving The Columns and keeping the um baselines low so the T the uh kind of technical detail is that the injector the column and the detector need to be here thermostated separately because uh the column temperatur start from very low temperatures where the while the injectors Ed to be uh need to be quite High temperatures um so this is a a technical need to have them separately separately controlled and uh it's also important that when
we use temperature programs the liquid phase that we use as a stationary phase would be stable over the whole temperature range that we use for the chromatographic analysis as well as that it has sufficiently low viscosity also at the lower temperatures where we start our chromatography so This means that the earlier Peaks would not have somewhat less efficiency due to the slow down mass transfair in the stationary effect so this is also important now when we have our compound separated and carried out from the column we also need to detect them somehow to to see
what columns were there and also to quantify them and some of the most common detectors in cast chromatography are the flame ionization Detector and also Mass spectrometry but traditionally also electron capture detector is popular but there are many many other te detectors and just to give you a short um kind of Link there are also termal conductivity detector which is kind of um let's say least selective detector um also nitrogen phosphorus detector to specifically detect compounds containing nitrogen or or phosphorus as well as for uh obtaining somewhat more structural Information coupling of GC with infrared
uh spectroscopy is possible let's look at the the some of the most popular detectors one by one so the kind of golden standard in gas chomatography is the flame ionization detector um the idea of the flame ionization detector is that the uent from the column is mixed with a hydrogen which is later mixed with an ear and this a mixture is lit in fire so that the hydrogen is burning in the as a Mixture with the air so we have a flame here and in these Flames ions are produced from compounds that contain carbon uh
so carbon containing compound somewhat pury in this flame and produce canons which cause conductivity in this flame and to measure this conductivity there is a collector electrod just on on top of the of the of the burner of the flame and uh we measure the conductivity as the as the uh real from The Collector Electrode relative to the burner tip so the conductivity is measured between these two two electrodes The Collector electrode up on on top of the um flame and then the tip from the from the uh flame carrier um now because the compounds
that cause the conductivity are compounds which were kind of paralized in this oxygen hydrogen flame and they have to contain carbon so all compounds that contain carbon on almost are giving A signal in the flame ionization detector and this is also the sole source why it is so widely used and why it's kind of a golden standard for gas chromatography analysis uh generally the response is proportional to the number of carbon High atoms so um ethane um has twice um larger signal or response factor in flame anization detector than does Methane uh but heter atoms
such as oxygen nitrogen decrease this signal so For example CO2 does not give any signal in the flame ionization detector um but this is kind of an exception also for example ammonia which contains only only um only nitrogen and hydrogen doesn't give any signal in the in the flame ionization detector the same for example for water but compounds that do contain carbon contain carbon carbon and carbon hydrogen bonds they do give a signal and you roughly said it's proportional to the number of carbon atoms but oxygen And nitrogen bonds with carbon decrease this response this
means that in order to do quantitative analysis to get good quantitation we need actually analytical standards to construct the calibration graphs and to quantify the compounds because the response is not directly proportional to the um carbon number there are small variations uh the generally the detector temperature should be sufficiently high so that the compounds that elude from The column would not um kind of precipitate or or condense in the detect s uh in case of flame ionization detector it's also additionally important that the water that is being formed during the burning in the flame would
not condense so it has to be at least 125 degrees but usually it is somewhere above 250° um it is very sensitive so the LOD values toe to bpb level can be achieved Of course compounds which have more carbons they are more for this the flame ionization detector is uh more sensitive it also has a very wide uh linear range up to six orders of magnitude which is ideal for quantification purposes it's also very robust and stable so there aren't much of a variations it's all the flow rates of the gases are kept constant and
um closely uh closely uh closely reducible flows of the gases Are achieved and it is suitable for almost any organic compound that contains carbon carbon carbon water car carbon uh hydrogen bonds and uh also all carrier gases are possible so nitrogen the helium of course H but also hydrogen can be used though as said before hydrogen is not really suggested due to its uh dangerous properties um now some compounds also contain other uh other atoms that could Um give advantageous DET detection properties for example classically pesticides contained a lot of halogenated uh hogen molecules and
hologen molecules have actually another advantageous property for detection and this has been used in the electron capture detector to get es especially low detection limits for compounds containing halogens uh namely the hallogen atoms are able to capture electrons so um so Attach electrons from the G from in during collisions with the electrons essentially and what happens in the electron capture detector is that we have a radioactive source so uh nickel 63 which is a source of beta particles and these beta particles can um react with the nitrogen as a which is used as a carrier
gas and um with the during the Collision uh electrons are emitted from the nitr so therefore we we have here in this Part uh electron Rich medium and when our unalike molecules are coming with the carrier gas to this detector then they capture the electrons and with this they re they reduce the conductivity in this medium and uh this conductivity is measured by measuring the conductivity between the cathode and the anode here so analyes decrease the conductivity and um here comp any compound that is able to capture the electrons can be detected and this this
Means largest sensitivity for the halogenated atom containing compounds the drawback of the elron capture detector is the the radioactive source that is used here and um it is also not so robust so any kind of contamination uh can build up here and uh cause um instability from Tay to T from sample to sample however if we are having galtes which contain hallogen atoms then extremely low detection limits can be achieved Even 10 time even uh even lower than with um flame ionization detector are possible and it also depends on how many halogens are uh present
in the molecule so the higher the number of hallogen atoms the more sensitive um analysis can be performed for these compounds the linearity is usually lower than for flame ionization detector so around three orders of magnitude and also the SEL the sensitivity depends on which of the hogen molecules atoms is present in The molecule so I iodine jber iodine the sensitivity is the best and lowest for the fluide um the stability of the instr of the electron capture detector is not as that it is for the flame ionization detector so contamination is a big problem
um and it's very important that the nitrogen that is used as a carrier gas is Ultra Pure to get um nice stable um conductivity lastly um you will actually Have a specific lecture about Mass spectrometry later uh but mass spectrometry is also a very important detector in both gas chromatography and liquid chromat graphy and it was first used also of course for for gas chromatography and usually we use something which is called electronization Mass spectrometry uh which is able to detect almost all compounds uh but the limits of detection highly depend on the Compound and
it can it is compatible very or competible with um detection limits in flame ionization detector so it is in the ppb range or for some compounds it can even be in the PPT range the linear range is somewhere between the linear ranges of the electron capture detector and the uh flame ionization detector and the sensitivity is increased for compounds which have polarizable um functional groups so Aromatic compounds or some fun functional group such as chlorine or ammonium group they all improve the sensitivity of the mass spectrometric analysis however the largest uh advantage of mass spectrometer
as a detector in gas chromatography is that um um it is kind of selective in the sense that we not only register the retention time of the compound and the peak area but we also uh obtain Mass Spectre of the compounds Which allows us to at least tentatively identify the compounds or get some additional Clues what compounds they could be even if we don't maybe have the analytical standards in the lab or we aren't directly um aiming to analyze um aiming to SP to um quantify specific compounds we can also look at the other compounds
uh that we have that have been present in our our sample so for example if the peppermint oil characterization is done then we could look at each of The Peaks and see what is the mass Spectra behind this peak and then use the Spectre to identify the compounds um and it's not only that um the Spectra as such can be analyzed by the analyst but there are also nice libraries available for the gas chromatography mpect gal is for example one very famous one is the n Library which is almost built in into all of the
into all of the GC Ms analysis and this is very convenient to get some Clues What compounds were present in the sample U when you also want to look at other compounds than the ones for which you have analytical standards already already there so now to kind of sum it up but also give you perspective into the limitations um the analyes that we are determining I said many times until now they have to be volatile so generally we can predict which one which ones of them can be analyzed based On the boiling points we can
also generally say which of the compounds would elude earlier and which would delute later uh based on these boiling points and they need to be stable so uh the compound should not degrade at the temperatures that we need to use for the chromatographic analysis and the same is valid also uh for the solvent that we use and solvent additive so we should actually use as pure solvent as possible so that there would be as little Impurities as possible the impurities of the solvent causes Coast Peaks so Peaks that don't actually belong to a sample uh
so the solent pur is also very important uh now let's look how the boiling point of the compound uh determines on is um associated with the structure of the compound and firstly let's look at the row here so we have here from C4 to C8 to C12 and we see that if ban is actually a gas that room temperature its boiling Point is minus1 De in Celsius then the C8 Octan is already having a boiling point of 126 degrees and D um dakan so C12 has even the boiling point of 216 still all of these
alkanes uh from C4 to C12 can be uh determined with the gas chromatography so the temperature range of their pointing points is sufficiently um yeah the temperature are not too too high however uh here temperature program would of course be useful to actually Get a good separation of all of these three alkanes or maybe also the alenes that have not been indicated here but which are uh with a intermediate number of carbon atoms in between them however when we start adding uh Clarity so for example when we look at the monocarboxylic acid of C8 then
we see that even though we didn't add any carbon atoms but we did have a add um carboxilic acid group here the boiling point Rose dramatically so we Have a boiling point more than 100 degrees in celsius higher and if we actually look at the C8 dicarboxylic acid then here the temperature has Rose dramatically to even uh more than uh 450 degrees and these uh temperatures are too high to be actually chromatographed nicely so for example the analysis of such um very polar molecules or molecules with very polar groups such as the um dicarboxylic acid
here is directly impossible with the gas Chromatography due to the temperature limitations however to overcome this derivatization is very widely used in gas chromatography and this derivatization is uh the reaction of the analy to change properties so that it would be possible to be analyzed with a gas chromatography and for the carboxilic acid acids this reaction is very often etherification so for example methylation of the um carboxilic acid groups to the corres bonding Methylesters and we can see that here if we would methylate these carboxilic acids we would be able to decrease the boiling point
of the monocarboxylic acid by about 40° but the effect is even larger for the die acid where we can actually now reduce the temperature to 2 uh 68° so that this compound now becomes also analyzable with gas chromatography so therefore for um for example FTI acid analysis Derivatization via esterification is a very common tool to enable cast chromatography analysis um we don't do only quantitative analysis with gas chromatography we largely or maybe today even mostly do also quantitative analysis and um quantitative analysis exclusively is done in gas chromatography with a calibration graph method uh due
to the fact that response factors with any of the common detectors Are not uh the same for all of the compounds and they are also not directly uh obtainable from the from the structure of the compounds so therefore calibration graph method is the most um advantageous way to quantify the compounds uh addition due to the fact that injection as well as the split uh can be somewhat not so good reproducibility internal standard method is um advantageous in the gas chromatographic Quantification and du and these small variations in the injected sample volumes can very nicely be
accounted for with the uh internal standards that have been spiked into the samples so with this the gas chromatography analysis can also very nicely be made quantitative so this was an introduction to the gas chromatography uh which is then primarily the for the analysis of volatile organic compounds or compounds That can be turned volatile with the derivatization uh in the next lecture we'll also look at how even more polar compounds can be analyzed with the liquid chromatography good luck








![Nobelpreis 2025: Verstehen wir Quantenphysik wirklich? [Ganze Doku] | Terra X Harald Lesch](https://img.youtube.com/vi/QbyKWvjIhP8/mqdefault.jpg)