my name is Russell DuBose Boyd and I'm from the department of molecular genetics at the University of Texas Southwestern Medical Center in Dallas Texas in this presentation I'll be talking about the feedback regulation of HMG co-reductase which is the rate-limiting enzyme in the synthesis of cholesterol so this slide shows a structure of cholesterol anaemic characteristics of this important molecule cholesterol is a sterile which is distinguished by this four ring structure this four ring structure in part stiffness on this molecule which makes it an ideal component of cell membranes now because cholesterol has a large number
of carbon carbon and carbon hydrogen bonds this molecule is virtually insoluble in water so because of this reason cells must be able to maintain cholesterol in a narrow range such that enough cholesterol is produced for the Sailor needs in the molecule but avoid the toxic over accumulation of cholesterol over accumulation of cholesterol can be toxic at the cellular level now this slide shows some of the essential functions of cholesterol cholesterol is absolutely necessary for life as I mentioned earlier one of the most well recognized roles for cholesterol is its role in cell membranes where it
maintains optimal membrane fluidity now cholesterol turns out to be a important precursor for very important molecules such as steroid hormones which help distinguish between girls and boys bile acids which aid in digestion and nutrition by solubilizing dietary fats and fat soluble vitamins and then finally cholesterol is abundant in the brain where it's found in myelin sheaths that surround axons and help in sunette synaptic transmissions now cells in our bodies mammalian cell million cells acquire cholesterol through two sources one of the sources is illustrated in this slide and as through the synthesis of cholesterol from the
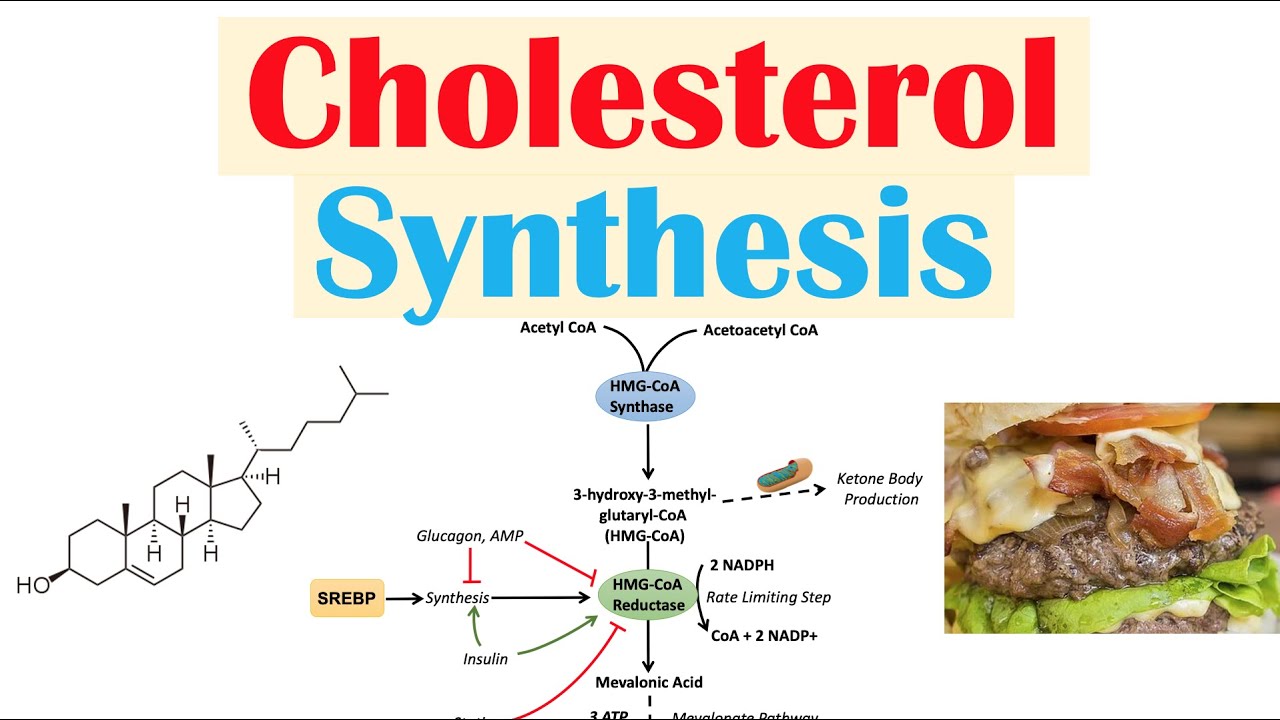
precursor acetyl co a now the conversion of acetyl co a to cholesterol occurs to the action of more than 20 enzymes now as you can imagine the synthesis of cholesterol involves the production of several intermediates and these intermediates themselves can also be converted to very important in products for example this compound farnesyl pyrophosphate is a precursor for an important compound called tilde call which is involved in in link like oscillation it's also a precursor for heme and ubiquinone x' which are involved in cell respiration vitamin k which is involved in blood coagulation and then finally
this farnesyl pyrophosphate and general general pyrophosphate become attached to many signaling proteins small GTP proteins directs them for membranes and this modification is absolutely necessary for normal cell function now in this slide shows that the synthesis of cholesterol occurs in various tissues at different rates now the liver and the Drina glands synthesize the most cholesterol in our bodies and i should point out that this was actually done in mice but similar effects are seen in humans and other primates the liver the liver synthesize large amounts of cholesterol for mainly production of lipoproteins and also for
bile acid synthesis the adrenal glands produce cholesterol primarily for steroid hormone synthesis whereas the gut synthesize cholesterol to force for cell division a large number of cells in the gut slough tauf every day and must be replaced by new cells which require a market amount of cholesterol synthesis should also point out that the gut is also a source of lipoprotein production so now the second source of cholesterol is actually from the lipo proteins that are produced by the liver and the intestine so shown here is a model of the low-density lipoprotein this is a major
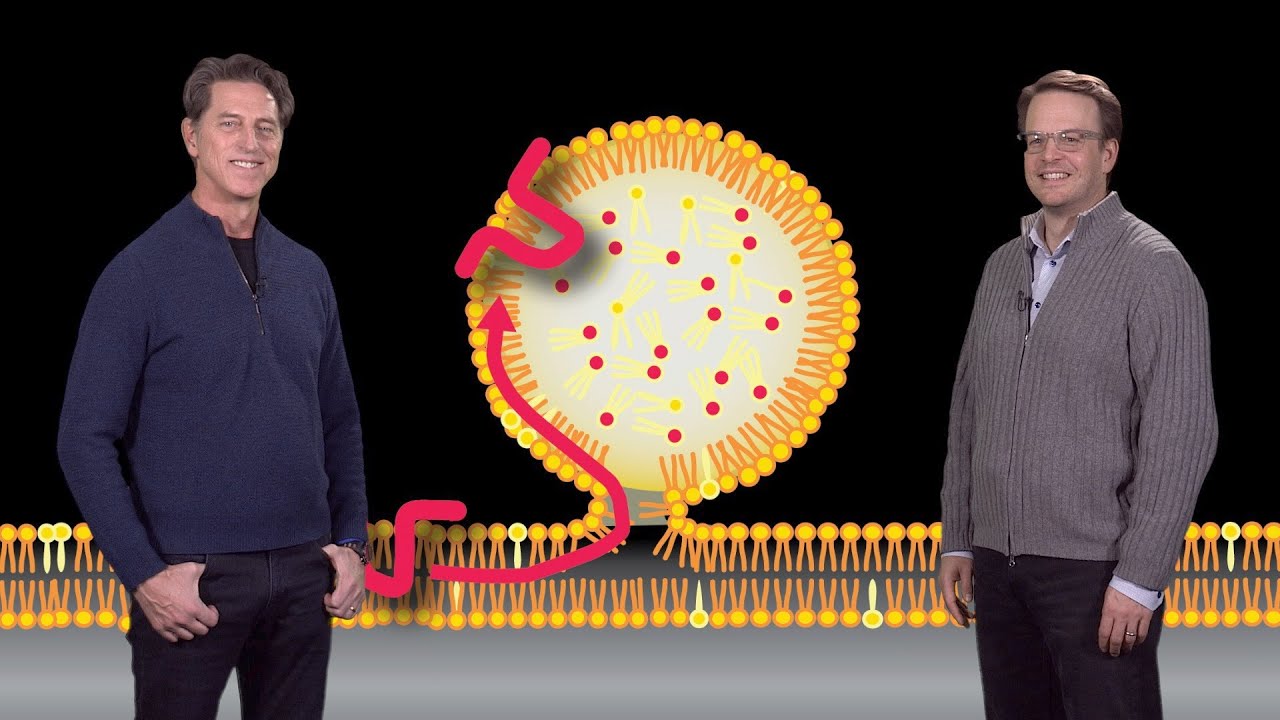
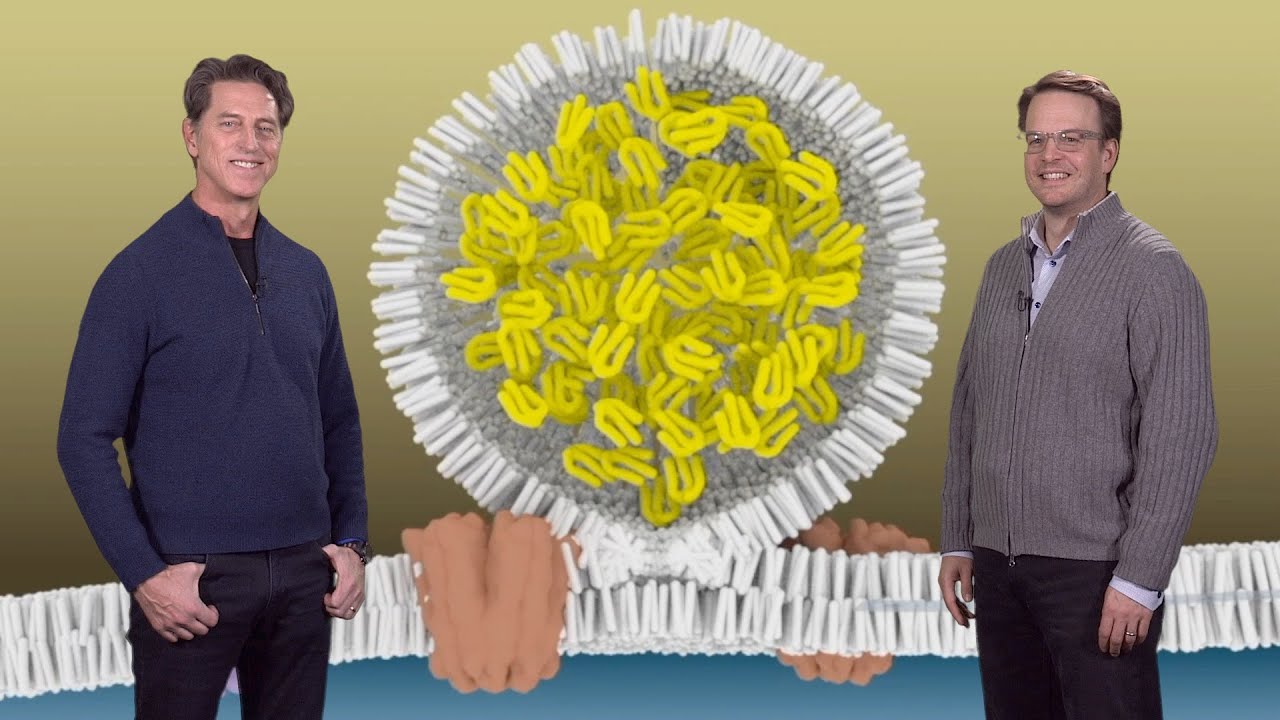
cholesterol carrier in human plasma so the low-density lipoprotein or LDL consists of a core that's composed of free cholesterol so the hydrophobic cholesterol forms the core of the LDL particle now this hydrophobic cholesterol is surrounded by a shell that's composed of a phospholipid with various amounts of a steroid cholesterol this is cholesterol to which a fatty acid has been attached to it that's intercalated into the phospholipid shell now this entire LDL particle is surrounded by a protein called a polite protein B so this light actually depicts how cells acquire cholesterol from these two sources endogenous
synthesis and from LDL so L do accept receptors there on the surface of cells bind to LDL by interacting with this a pillai PO protein B particle that surrounds the lipoprotein shell once the LDL particle binds to LDL receptor the entire complex is internalized into coated pits and these coated pits are then targeted to lysosomes where the LDL particle is degraded in the cholesterol free cholesterol is now liberated and provided to the cell for various uses so again these two sources of Sayre cholesterol either from the receptor or LDL receptor mediated endocytosis of LDL or
through endogenous synthesis can be used interchangeably so for example if elio becomes limiting the cell switches to endogenous synthesis for sources of cholesterol and if the endogenous synthesis is blocked then the cells can now use exogenous LDL for their source of cholesterol so we've talked about the essential function of cholesterol it's important in cell membranes it's an important precursor of steroid hormones and bile acids however there's a bad side of cholesterol and that's illustrated in this slide for a number of years elevated levels of blood LDL cholesterol is associated with a risk for coronary heart
disease and heart attacks so shown here you can see that the level of blood cholesterol literally Corrie correlates with risk for coronary heart disease and this is because elevated cholesterol can actually deposit in the arteries that lead to the to the heart and over time this deposition results in a disease called atherosclerosis in which this deposition of cholesterol can lead to the production of plaques that ultimately can block blood flow to the heart thereby creating a heart attack now one of the most widely prescribed drugs to lower LDL cholesterol are a group of drugs called
statins shown here is the typical core structure of the statins and some of the various forms of statins that have been utilized in the clinic over the years as statins have become one of the most largest selling medication medications in the United States because their ability to lower blood LDL cholesterol so shown in this experiment is a summary of at least four studies that reveal that statins indeed reduce LDL cholesterol and that reduction leads to a reduced incidence of coronary heart disease so shown here in the closed circles are clinical trials in which patients were
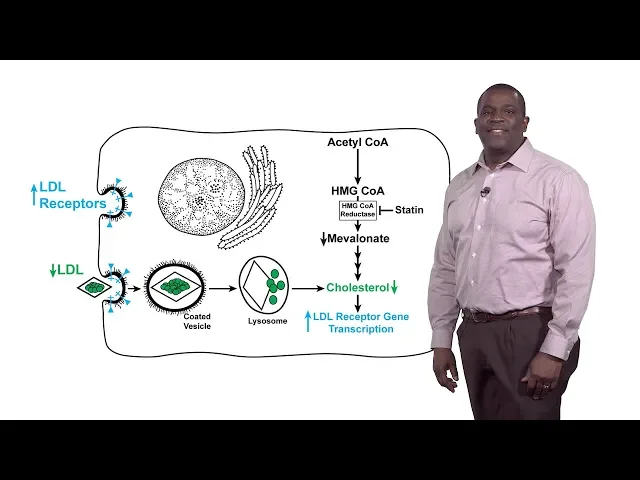
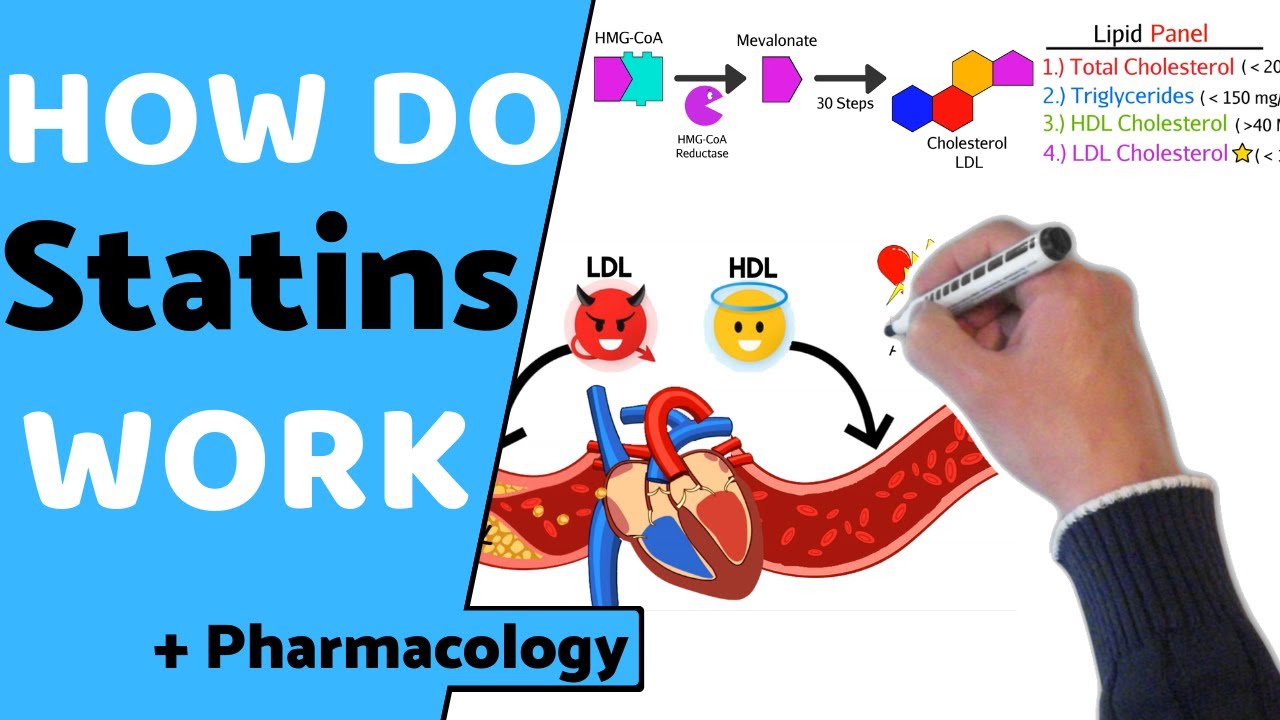
either treated with a statin shodhan shown in the closed circles or a placebo and in each of these studies the statin treatment led to a drop in LDL cholesterol levels and this drop in LDL cholesterol levels led to a reduction in coronary events ie heart attack so then the question becomes is you know how two statins work and what do statins do so we first answer what do statins do so statins inhibit the enzyme HMG co-reductase HMG co-reductase catalyzes the rate limiting step in the synthesis of cholesterol it's actually the fourth step in the cholesterol
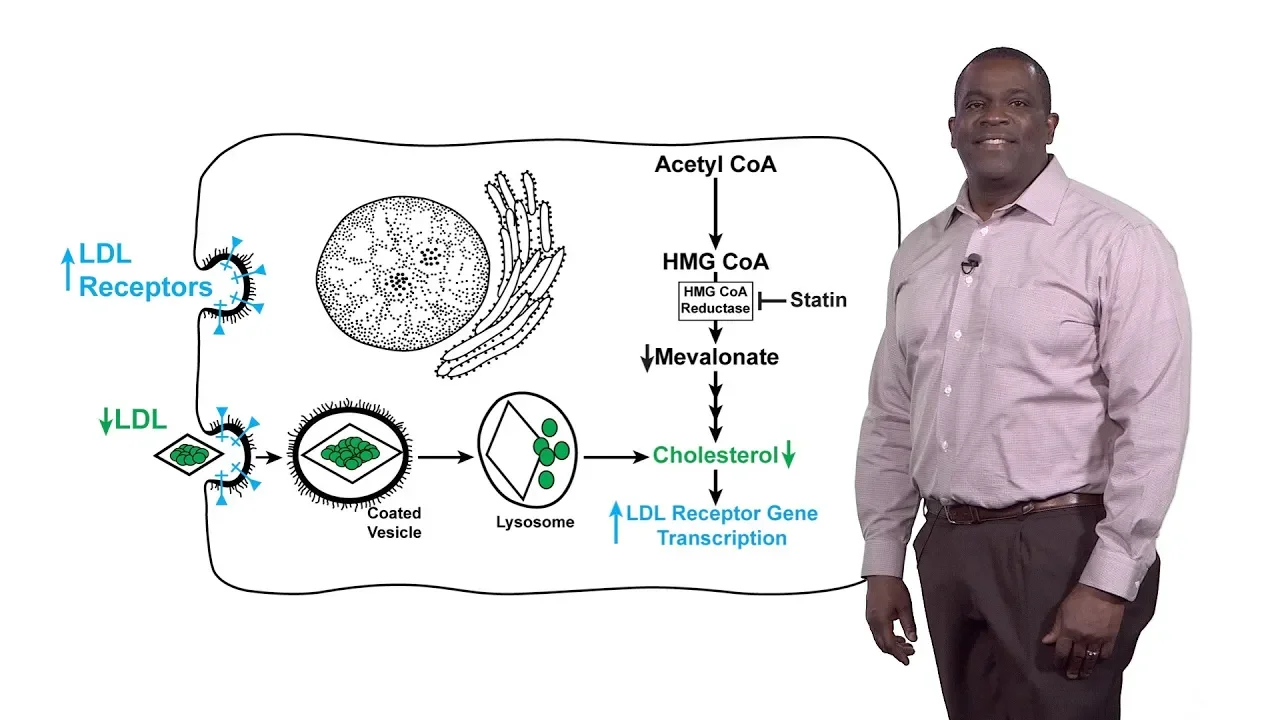
synthetic pathway so statins competitively inhibit HMG querida taste by mimicking the product of the reductase reaction metal on a so it's competitive inhibition of the reductase underlies the ability of statins to down regulate LDL cholesterol in the blood so how do statins work so again by competitively inhibiting HMG co-reductase this leads to a drop in the amounts of metal on eight and of course a drop in cholesterol this cholesterol depletion leads to an increase in the transcription of the gene encoding the LDL receptor and as a result the number of the LDL receptors on the
surface especially of the liver cells and this increased LDL receptors leads to an increased or enhanced a uptake of blood LDL and that reduction in blood LDL is responsible for the lowering of coronary heart disease in statin treated patients however the clinical effects of statins are actually blunted by the compensatory increase in HMG co-reductase at a company statin therapy and that's illustrated in this slide this is an immuno blot of HMG co-reductase protein in the livers of mice that have been fed a statin or even in cultured cells that have been treated with statins in
vitro when as you can see statin treatment causes a marked accumulation of HMG co-reductase in this accumulation as I mentioned earlier blunts the clinical effects of statins so our next question is why do statins cause HMG co-reductase to accumulate to such a high level which I should point out has been estimated to be at least 200 fold so normally HMG co-reductase is subject to an enormous amount of feedback regulation in this feedback regulations meaning in part by sterols now statin treatment as I mentioned earlier blocks HMG co-reductase activity and it prevents the synthesis of these
sterile molecules and of course that prevention of sterile synthesis actually is responsible for the up regulation of the LDL receptors and subsequent reduction of LDL cholesterol however because the statins blocked the synthesis of sterols it disrupts the feedback regulation of the reductase and as a result three events happen first because of this reduction in cholesterol and other products of the cholesterol synthetic pathway there's an enhanced transcription of the reductase gene there's an enhanced translation of the reductase mRNA and then finally there's an enhanced ability of the reductase protein so these three events are responsible for
that marked increase in the reductase protein I showed you in the previous slide so over the years my laboratory has been interested in trying to understand the molecular mechanisms that underlie this enhanced ability to protein and that will be the subject of the remainder of this presentation so this slide illustrates that sterols indeed accelerate the degradation of HMG co-reductase so in this experiment we used classic pulse chase analysis to monitor the stability of reductase in cells that have been treated in the absence or presence of sterols so what we do here is we typically label
cells with radioactivity a small subset of HMG co-reductase molecules we then take that radioactivity away and then follow the press the stability of the reductase protein in the absence or presence of sterols and as you can see here when the cells are chased in media that contains no radioactivity in the absence of sterols the reductase is fairly stable over time however you can see the addition to sterile in the chase medium causes the reductase levels to markedly diminish again this is indicative of the sterile accelerated degradation of HMG co-reductase now to understand the molecular mechanisms
for the sterile induced degradation through duct taste we've got to understand the structure of the HMG co-reductase protein in the domain structure of the reductase is actually illustrated in this slide so HMG co-reductase consists of two distinct domains it has an in terminal domain that anchors the protein in the membranes of the endoplasmic reticulum or the ER now this internal domain which we refer to as a membrane domain contains eight membrane spanning regions and is followed by the second domain of HMG co-reductase which we call the catalytic domain so the catalytic domain protrudes into the
cytosol of cells and it contains all the enzymatic activity of HMG co-reductase in fact a truncated version of the reductase it only contains the catalytic domain can completely rescue the synthesis of mevalonic in cells that lack HMG co-reductase so the catalytic domain is both necessary and sufficient for the synthesis of mevalonic which then raises a question why is this protein membrane bound and it turns out that the reductase is actually a membrane-bound protein as far back as its yeast so the function of the membrane domain of reductase was illustrated in this early experiment that compared
the stability of the catalytic domain which remember contains all enzymatic activity to the full-length enzyme and again a simple pulse chase analysis was used to monitor the stability of these two proteins as you can see in the panel on the left the truncated catalytic domain produces a very stable protein that importantly its degradation is not influenced by sterols in contrast the full-length protein which again contains the membrane domain is less stable even in the absence of sterols and you can see that sterols markedly accelerate the degradation of HMG co-reductase which indicates that the membrane domain
the function of the membrane domain is for this sterile accelerated or sterile induced to degradation so what the previous studies indicated is that the membrane domain of a reductase is necessary and sufficient for sterile accelerated degradation and is suggested that the membrane domain either directly or indirectly consents intracellular levels of sterols and the sensing results in perhaps a conformational change in the reductive membrane domain that causes a protein to be susceptible to rapid degradation and of course because statins block the synthesis of cholesterol statins indeed block this will we call er associated degradation or --red
of HMG co-reductase so key breakthrough in our understanding of the erat of HMG co-reductase came with a discovery of a pair of proteins ER membrane proteins called insig one in in situ these insig proteins for the purposes of this talk are very redundant they perform redundant roles in the degradation or erat of HMG co-reductase they're identical they're about 85% identical and they're highly hydrophobic proteins now the role of n6 in the erat of HMG co-reductase was first illustrated in this experiment so here again we use pulse chase analysis to measure the sterile accelerated degradation of
the reductase in cells that were either transfected with control molecules called SI rnas or cells that were transfected with SI rnas that would lead to the knockdown of expression of both in cig 1 and NC 2 and as you can see in the panel on the left and the cells can transact it with the control SI rnas sterols markedly accelerate the degradation of HMG co-reductase so open circles or experiments conducted in the absence and the closed circles are can experiment conducted in the presence of sterols and what you'd readily see is at the knockdown of
in cig 1 and n cig to completely abolish this sterile accelerate degradation indicating that these proteins play a key role in the process so our next question is what is the mechanism by which in 6 accelerate the --red of HMG co-reductase so this experience slide shows that inhibitors of the proteosome 26s proteosome block the sterile induced degradation of HMG reductase so as you can see in this experiment first two lanes sterols caused the reductase to become markedly degraded and this degradation is completely blocked when these cells are treated with the proteasome inhibitor so with this
allows us to create another model in which again the membrane domain of the reductase either directly or indirectly senses levels of intracellular sterols this causes the reductase to bind to in sig's and that insig binding leads to reactions that caused the reductase to now be degraded by the 26s proteosome now it's known that most substrates of the Proteas ohms require their prior ubiquitination ubiquitination is a process by which the small protein ubiquitin becomes covalently attached to substrate molecules and once a chain of ubiquitin ZAR attached to substrates it becomes recognized by the Proteus ohms for
degradation now this is called poly ubiquitination and poly ubiquitinated proteins requires the action of at least three different types of enzymes that's illustrated in this slide in the first step ubiquitous becomes activated in an atp-dependent manner by an enzyme called II 1 or ubiquitin activating protein in the next step the ubiquity is transferred from the e 1 to the 2 another enzyme called e 2 or ubiquitin conjugating enzyme in the final step the e 2 combines with an e3 or ubiquitin ligase which in turn is associated with the substrate shown here in green what the
e 3 does is facilitates the transfer of ubiquitin from the ubiquitin conjugating enzyme to a lysine residue in the substrate protein generating a ubiquitinated substrate now this process occurs many times until a ubiquitin chain is built upon the substrate protein that can now be recognized by the proteasome for degradation so considering that these instinct proteins are required for the degradation of the reductase and that the reductase actually is degraded by Proteus ohms our next question is is reductase ubiquitinated that question was answered in this experiment shown in this slide so we've done this experiment is
we've treated cells in the absence and presence of sterols and the proteasome inhibitor after these treatments we immunoprecipitate the reductase and then probe those embryo precipitates for either total reductase shown on the bottom panel or ubiquitinated reductase so as you can see in the first Lane even though reductase is pulled down in these experiments we see no reactivity with ubiquitination however sterols cause the reductase to become ubiquitinated and this ubiquitination is markedly increased if we also include proteasome inhibitors it sort of indicates to us is that steroids indeed cause the reductase to become ubiquitinated and
this ubiquitinated protein is now degraded by Proteus ohms as indicated by the stability of ubiquitinated reductase by these proteasome inhibitors our next question is insects required for this sterile induced ubiquitination of the reductase and again we turn to si RNAs cells were transfected either with the control si RNA or s RNAs against n cig 1 and 2 we then treat these cells in the absence of presence of steroids and then probe for ubiquitinated reductase as you can see in the first two lanes the reductase is nicely ubiquitinated in the presence of sterols and that knockdown
of in Sigma 1 and in stick to completely abolish this ubiquitination so that now allows us to fill more gaps in our model for the in cig mediated erat of hmg-coa reductase now it turns out that a subset of in cig molecules actually associate with an e 3 e to ubiquitin ligase complex again in the presence of sterols the membrane domain of reductase senses the sterile and this sensing causes the reductase to bind to n6 and of course these these in six then bridge the reductase to the III II to ubiquitin ligase complex this bridging
then results in the ubiquitination of reductase at two lysine residues in the membrane domain and this ubiquitination then causes the reductase to be removed from the ER membrane and subsequently degraded by Proteus ohms through a process that we're not completely through processes not completely understood so in summary I've told you today that HMG co-reductase is a rate-limiting enzyme in the cholesterol synthetic pathway and it's a target of these cholesterol-lowering statin drugs the reductase is controlled through a very complex feedback regulatory system this mediated by cholesterol and other types of sterols and it's statins disrupt this
feedback regulatory system in part by blocking this insig mediated ubiquitination and --red of HMG co-reductase you